
DRUGAI
今天为大家介绍的是来自Daniel J. Diaz团队的一篇论文。工程化稳定的蛋白质是工业和制药生物技术发展中的一项基本挑战。本文介绍了Stability Oracle,一种基于结构的图transformer框架,该框架在准确识别热力学稳定突变方面达到了SOTA性能。作者的Stability Oracle框架引入了几项创新,以克服数据稀缺和偏差、泛化能力和计算时间等著名挑战,例如:通过热力学排列进行数据增强,使用结构氨基酸嵌入来对突变进行单一结构建模,蛋白质结构特定的注意力偏置机制使transformer成为图神经网络的可行替代方案。作者提供了训练/测试划分,以减少数据泄漏并确保模型评估的正确性。此外,为了检验数据工程的贡献,作者微调了ESM2表示(Prostata-IFML),在基于序列的模型中达到了SOTA。值得注意的是,尽管Stability Oracle在预训练时使用的蛋白质数量少了2000倍,参数量少了548倍,但其性能仍优于Prostata-IFML。作者的框架为微调基于结构的transformer以适应几乎任何表型奠定了基础,这是加速蛋白质生物技术发展的必要任务。

预测和理解氨基酸替换对蛋白质热力学稳定性(ΔΔG)的影响能力是开发蛋白质生物技术的核心任务,例如工业生物催化剂和制药生物制品。热力学稳定性增强的蛋白质不易展开和聚集,更易于工程化;稳定蛋白质的骨架可以进一步探索可能改善目标功能的潜在不稳定突变。热力学稳定性通过原生态与展开态之间的Gibbs自由能变化(ΔG)来测量,反映了整体结构的基本完整性。蛋白质工程是一个非常繁琐的过程,制约了蛋白质生物技术的发展,因此开发能够准确预测点突变ΔΔG并识别稳定突变的计算方法成为一个高度活跃的研究领域。
深度学习正在革新许多物理和生物学科,其中AlphaFoldV2在2021年被誉为年度科学突破,掀起了一波深度学习结构预测工具的热潮。虽然已经报道了多种基于序列和结构的深度学习框架用于稳定性预测,但数据缺乏和机器学习工程问题阻碍了深度学习算法在蛋白质稳定性预测方面产生类似的革命性影响。
对过去15年发布的最先进(SOTA)计算稳定性预测工具的系统分析,突显了数据稀缺、变异、偏差、泄漏以及评估模型性能的度量标准不佳等关键问题,这些问题阻碍了有意义的进展。因此,研究界仍主要依赖基于物理的方法,例如Rosetta和FoldX,或浅层机器学习方法。目前的SOTA计算工具在发布时通常报告75-80%的准确率,但这些准确率主要反映了它们在识别失稳突变(destabilizing mutation)方面的表现,而失稳突变占测试集的大多数,且识别它们对于鉴定致病变异至关重要。然而,当第三方在数据集上评估其预测稳定突变的能力时,实际上只有约20%的预测是稳定的。尽管稳定突变自然较少,但对稳定突变的较差泛化能力已被证明是由于训练测试集之间的广泛数据泄漏和当前计算稳定性预测社区使用的训练集中的严重类别不平衡(失稳突变占>70%)所导致的,这些问题需要解决以便于机器学习指导的蛋白质工程。最后,目前的计算工具主要用皮尔逊相关系数和RMSE作为评估指标。由于训练和测试集中稳定突变(
模型部分
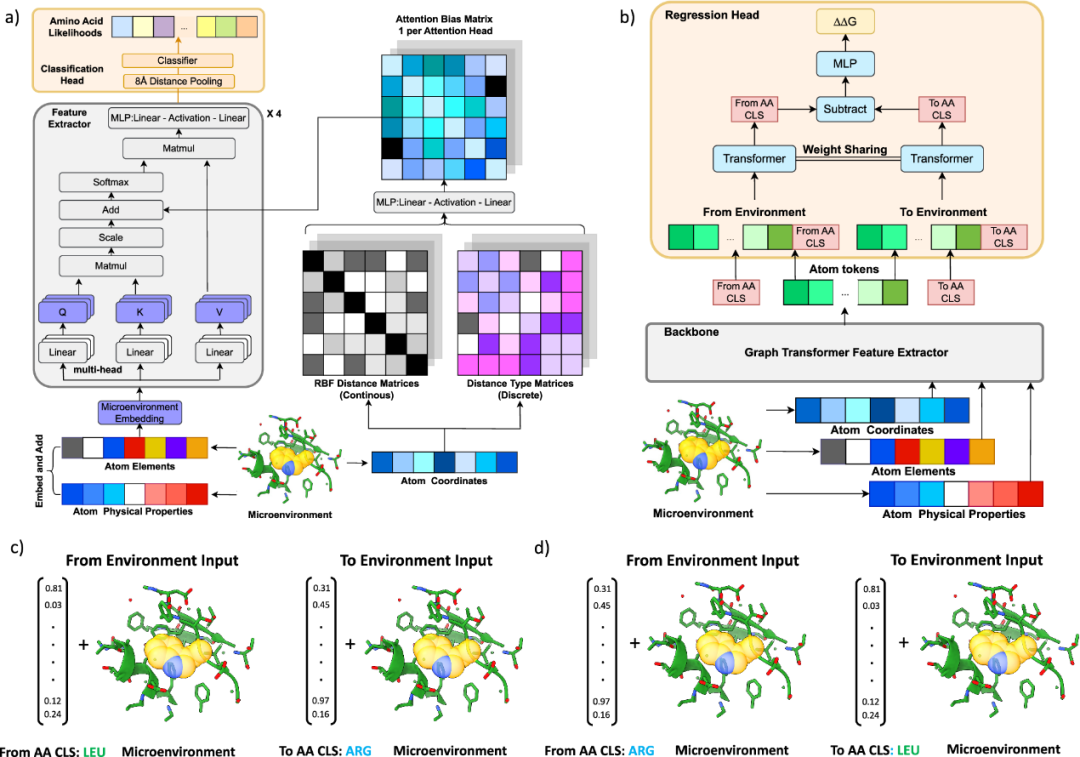
图 1
为了开发一个更强大且更具泛化能力的下游任务框架,作者首先构建了MutComputeXGT,一个图transformer版本的MutComputeX(图1a)。每个原子被表示为一个节点,具有原子元素、部分电荷和SASA值作为特征,并使用成对原子距离标记边。图transformer架构将成对距离转换为连续和分类的注意力偏置,为注意力机制提供基于结构的归纳偏置。为了生成掩蔽氨基酸的概率,作者对掩蔽Cα 8Å范围内所有原子标记的最终层隐藏表示进行平均。将池化范围缩小到掩蔽氨基酸第一接触壳内的原子的设计决策,是基于训练自监督3D CNN时系统变化微环境体积的见解。具有相似数量的参数和相同的训练-测试划分,MutComputeXGT展示了优于MutComputeX的表示学习能力,达到了92.98% ±0.26%的野生型准确率,而MutComputeX约为85%。
Stability Oracle架构利用MutComputeXGT的特征提取器和分类头进行监督微调(图1b)。以前基于结构的稳定性预测器需要两个结构(实验或计算得到的)来明确建模野生型和突变氨基酸。第二个(突变体)结构通常使用AlphaFold或Rosetta等计算技术获得。这种方法的缺点是:(1)计算方法在推理时变得昂贵,(2)难以评估计算得出的突变体结构的质量。相比之下,Stability Oracle不依赖于第二个结构。具体来说,从特定残基周围的局部化学环境(掩蔽微环境)中提取结构特征,并将突变表示为“from”和“to”氨基酸嵌入向量对。为了建模特定突变的ΔΔG,使用初始结构的微环境在回归头中对“from”和“to”氨基酸嵌入进行上下文化(如图1b所示)。这种架构设计使框架能够隐式学习“from”和“to”氨基酸如何与局部化学环境相互作用,而不依赖计算结构预测工具提供化学相互作用。“from”和“to”氨基酸嵌入来自MutComputeXGT分类器最后一层的权重。这个设计决策基于以下观察:这20个神经元的权重表示在归一化为概率分布之前,代表了微环境特征与20种氨基酸中的每一种的相似度。因此,它们是20种氨基酸的基于结构的上下文嵌入,是从蛋白质数据银行(PDB)中50%序列相似性表示中自监督预训练得到的,作者称之为结构氨基酸嵌入。
作者强调了Stability Oracle中回归头的几个设计决策。其中值得注意的是使用了Siamese注意力架构,将突变嵌入视为两个分类(CLS)token(图1b)。CLS token在自然语言处理(NLP)领域常用于捕捉下游任务的全局上下文。由于原子和氨基酸是不同尺度的化学实体,作者设计了回归头,使特定的微环境对“From”和“To”氨基酸级别的CLS token进行上下文化。一旦上下文化完成,这两个氨基酸CLS token会相互减去,生成一个突变隐藏表示,然后解码为ΔΔG预测。这个设计强制执行Gibbs自由能属性的状态函数,为热力学可逆性和“自我突变”(ΔΔG = 0 kcal/mol)提供了适当的归纳偏置。
热力学排列(TP)数据增强技术
Stability Oracle框架旨在泛化到蛋白质结构中所有位置的380种突变类型。历史上,这类模型的开发一直受到数据稀缺、偏差和泄漏的限制。为了解决这些问题,作者精心挑选了训练和测试数据集,并开发了一种称为热力学排列(TP)的数据增强技术。
众所周知,以前的研究中一个主要问题是训练集和测试集之间包含相似蛋白质(“数据泄漏”),导致泛化能力评估不佳。已有研究表明,在突变、残基或蛋白质水平上的训练-测试集划分会导致对验证集的过拟合,需要严格的序列聚类来确保泛化能力的正确评估。因此,作者基于MMSeqs2计算的30%序列相似性阈值创建了新的训练-测试集划分。首先,构建了T2837测试集,然后使用该测试集从剩余的实验数据中移除任何同源蛋白质,生成C2878训练集。同样的方法用于从最近发布的cDNA展示蛋白水解数据集#1的单突变子集中构建cDNA117K训练集(图2)。
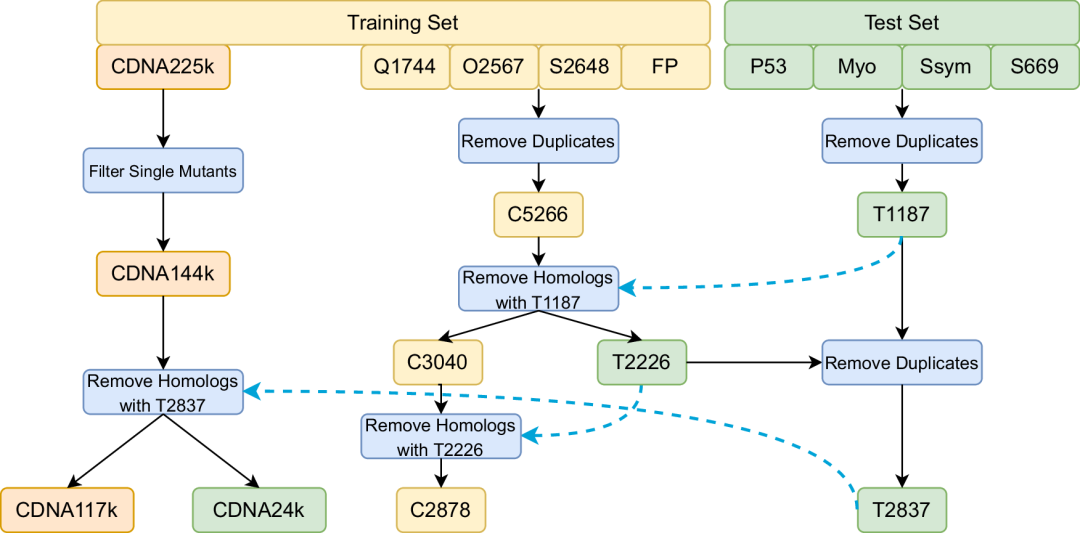
图 2
即使有了扩展后的T2837测试集,作者仍然无法评估380种突变类型中14%的泛化性能,因为这些类型未包含在T2837中。此外,T2837中的突变严重偏向于丙氨酸(图3a,底行),进一步阻碍了作者评估模型泛化能力的能力。研究界传统上依赖热力学可逆性(TR)数据增强技术来生成具有更多突变类型的数据集。然而,即使在C2878 + TR和T2837 + TR中,仍有约3%的突变类型缺乏数据。更重要的是,TR增强的一个主要缺点是它生成的所有稳定突变都是到野生型氨基酸,如图3b所示。这些突变在识别非野生型稳定突变方面没有预测能力,而识别非野生型稳定突变是蛋白质工程中热力学稳定性预测的主要目标。为了提高深度学习框架对稳定蛋白质的预测能力,需要额外的非“到”野生型氨基酸的稳定突变数据。
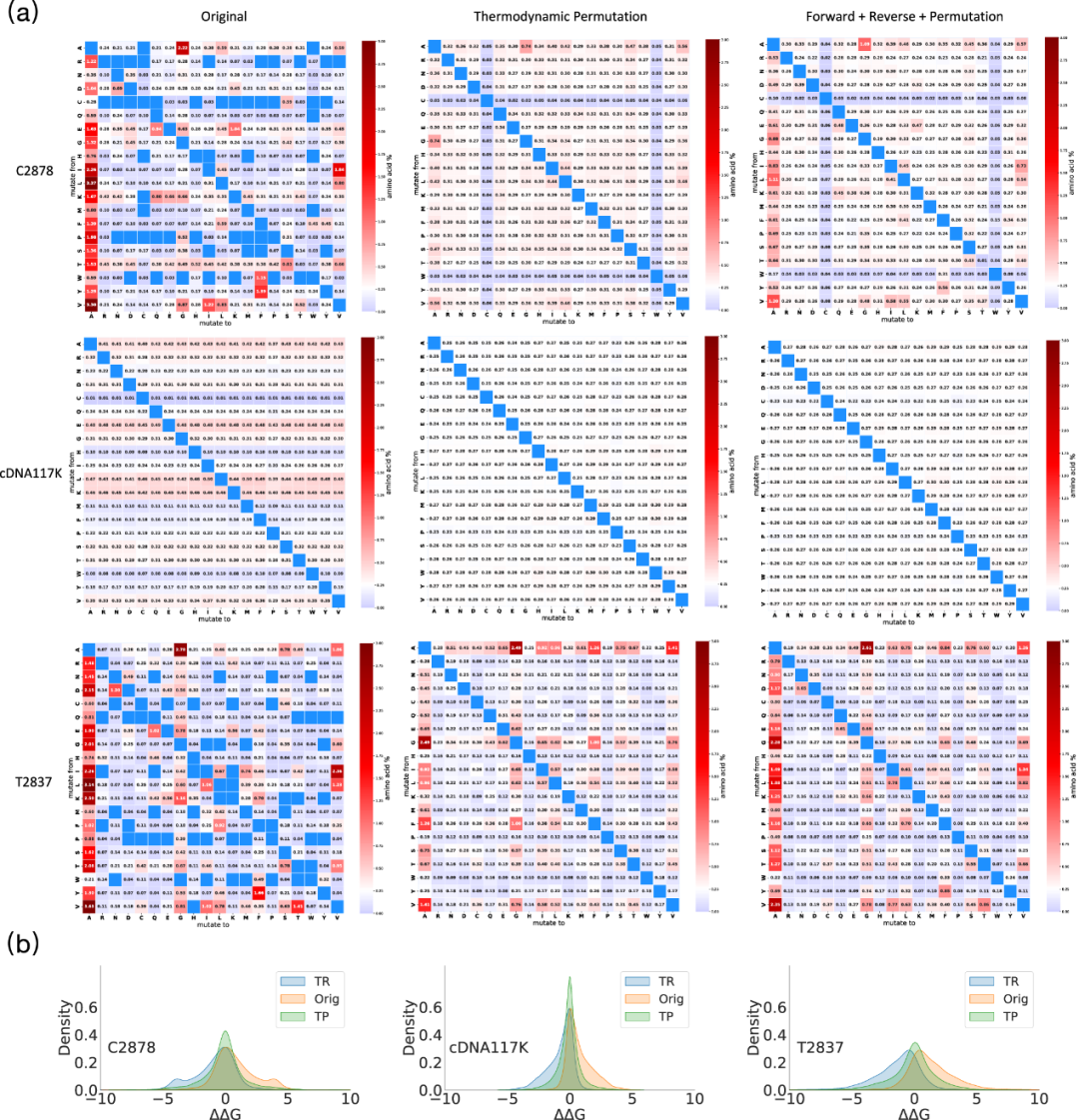
图 3
为了解决这些问题并提高Stability Oracle的泛化能力,作者引入了一种数据增强技术,称为热力学排列(TP)。TP基于Gibbs自由能的状态函数属性,可以在实验表征了多种氨基酸的残基处生成热力学有效的点突变。通过TP,作者能够分别为cDNA117K、C2878和T2837生成额外的202万、1.89万和7700个点突变,涵盖所有380种突变类型。此外,TP还减轻了所有三个数据集中的多种采样偏差(图3a,中列)。首先,TP为C2878和T2837中缺失的13.2%和14.5%的突变类型提供了突变数据。TP为C2878和T2837样本生成了380种突变类型的数据,提供了首个包含所有突变类型实验ΔΔG测量值的训练和测试集(cDNA展示蛋白水解数据集并不直接测量ΔΔG,而是从多重蛋白水解实验的下一代测序数据中推导出ΔΔG值)。
图3a展示了采样偏差的改进情况,红色(过度采样)和蓝色(欠采样)向白色(平衡采样)转变。在C2878和T2837数据集中,这在“到”丙氨酸的偏差中最为明显。在cDNA117K中,存在“从”丙氨酸、谷氨酸、亮氨酸和赖氨酸突变的过度采样偏差,而“从”半胱氨酸、组氨酸、蛋氨酸和色氨酸突变的欠采样偏差。TP完全平衡了cDNA117K突变类型分布,每种突变类型约占数据集的0.26%(100%/380),如图3a中列中行所示,均为白色。因此,cDNA117K的TP增强提供了第一个大规模的ΔΔG数据集(超过100万),均匀地采样了100个蛋白质域中的所有380种突变类型。与TR不同,TP不包括到野生型氨基酸的稳定突变,并产生了平衡的ΔΔG测量分布(稳定与失稳)(图3b)。
TP数据增强技术对模型性能的影响
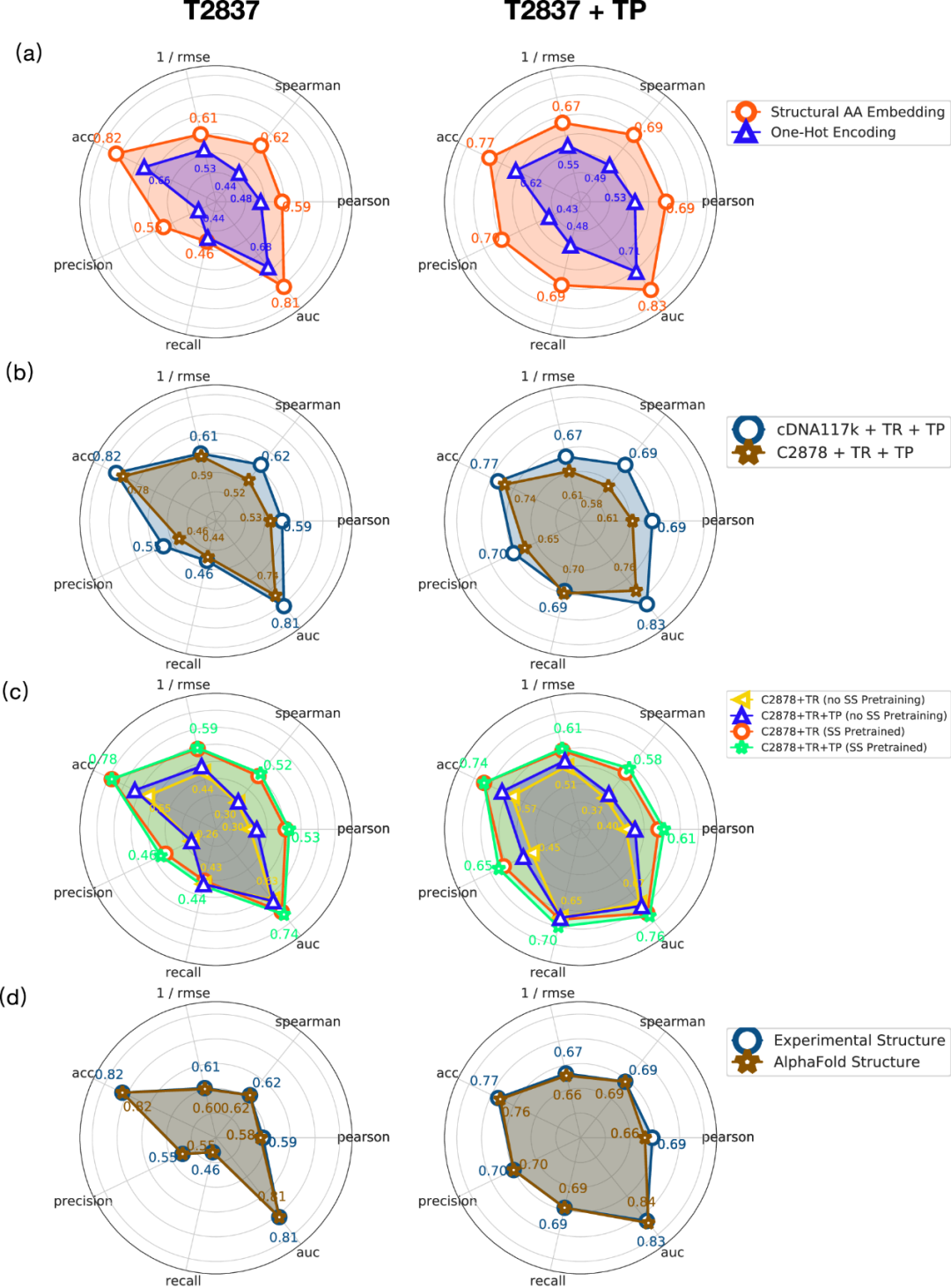
图 4
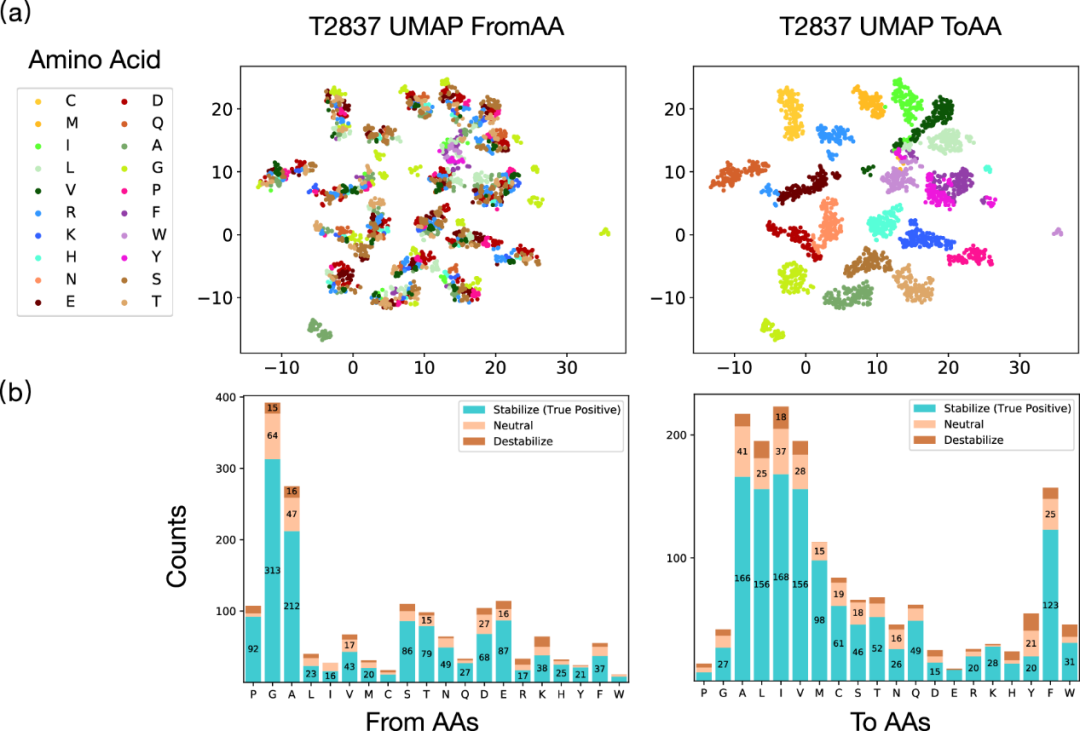
图 5
作者比较了在cDNA117K和/或C2878数据集上训练,是否使用TP增强,使用结构氨基酸嵌入和独热编码的效果,并评估了在所有测试集上的性能。作者观察到,结构氨基酸嵌入比简单的独热编码显著提高了性能(图4a)。对T2837的突变隐藏表示进行UMAP可视化显示,“ToAA”CLS token驱动了潜在空间的组织,并恢复了20种氨基酸之间已知的生化关系(图5a所示)。作者观察到:1)疏水性氨基酸(LEU、VAL、ILE、MET)、芳香族氨基酸(PHE、TYR、TRP)和短极性氨基酸(SER、THR和ASP、ASN)的聚类(右图);2)独特氨基酸(GLY、CYS、PRO)的隔离(右图);3)从GLY突变并添加手性侧链的独特情况(左图)。关于380种突变类型的特定残基案例研究。对于训练集,在cDNA117K + TP + TR上训练自监督表示在所有测试集上的回归和分类指标中表现最佳(图4b/c所示)。虽然这可能是由于其相对于C2878 + TP + TR的规模和突变类型平衡,但有趣的是,单域天然蛋白质的蛋白水解稳定性实际上通常是热力学稳定性的优秀替代指标(如原始出版物中所指出的)。
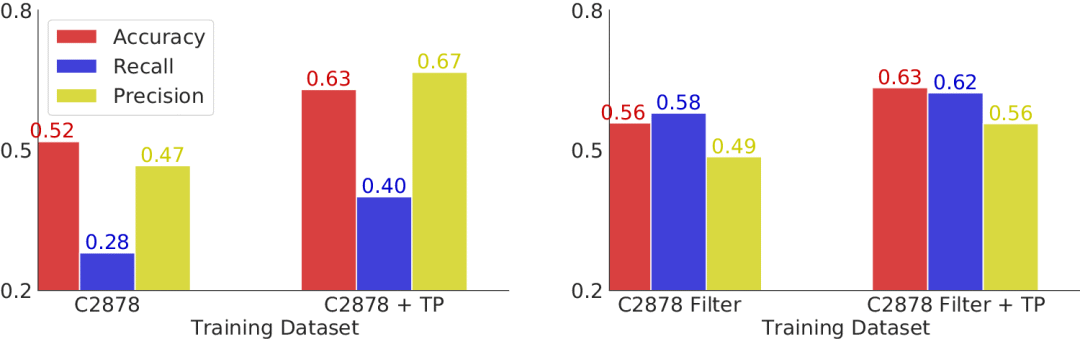
图 6
从这些数据中,TP对模型泛化的影响尚不清楚。为了进一步研究TP增强数据集如何影响泛化,作者评估了在T2837 + TP中突变类型的预测,这些类型在C2878 + TR中缺失但在C2878 + TP + TR中存在,即12种没有实验数据的突变类型。对于这些突变类型,TP提高了泛化能力:召回率从0.28提高到0.4,精度从0.47提高到0.67(图6)。作者将缺失数据的突变类型人为扩展到54种,观察到类似但减弱的精度和召回率的改进(图6)。
编译 | 黄海涛
审稿 | 曾全晨
参考资料
Diaz, D. J., Gong, C., Ouyang-Zhang, J., Loy, J. M., Wells, J., Yang, D., ... & Klivans, A. R. (2024). Stability Oracle: a structure-based graph-transformer framework for identifying stabilizing mutations. Nature Communications, 15(1), 6170.
