
DRUGAI
基于质谱的蛋白质组学旨在识别生成串联质谱图的肽段。传统方法依赖蛋白数据库,但在某些情境下受限或无法应用。de novo肽段测序无需先验信息,具有广泛的生物学应用价值,但因准确性不足而难以推广。研究人员在此提出InstaNovo,一种将碎片离子峰直接转译为肽段序列的Transformer模型,表现优于现有技术。进一步,研究人员开发了InstaNovo+扩散模型,通过迭代优化提升预测性能。该方法在多个数据集中实现更高的治疗性测序覆盖率,发现新型肽段,并识别未报道的生物体,拓展了蛋白质组学的搜索范围与检出能力,适用于直接蛋白测序、免疫肽组学及“暗蛋白组”等多个领域。

质谱(MS)技术革新了蛋白质组的研究方式。其中,主流的bottom-up蛋白质组学通过将获得的串联质谱(MS/MS)碎片离子谱与来自蛋白数据库的理论肽段碎片谱进行比对,实现肽段识别。目前普遍采用数据库搜索结合靶-诱饵方法评估错误发现率(FDR),以控制肽段和蛋白质识别的可靠性。
然而,这种方法存在局限:仅能识别数据库中已有的蛋白序列,难以发现剪接变体、单核苷酸多态性、外源蛋白或合成序列。此外,数据库搜索在考虑修饰时计算成本急剧上升,限制了可检测的翻译后修饰种类,也使开放搜索和半酶切搜索耗时巨大,并易引入较高的假阳性率。
作为替代方案,de novo肽段测序通过分析碎片离子指纹图谱,无需先验序列信息,是底层蛋白质组学的重要工具。尽管已有算法实现了一定程度的自动化,但仍存在FDR高、计算开销大等问题,限制了其在大规模实验中的应用。
研究人员在此提出InstaNovo,一种在de novo肽段预测中超越现有方法的新模型,显著提升了精准率和召回率。InstaNovo采用Transformer架构,利用多尺度正弦嵌入对MS峰值进行编码,并通过九层解码器进行交叉注意力建模,结合背包束搜索策略进行候选肽段筛选与打分。研究人员还进一步开发了InstaNovo+,一种受人工推理启发的扩散式迭代优化模型,可进一步提高预测准确性。
研究结果
模型架构与训练数据集选择
研究人员认为,Transformer架构若结合大规模、高质量的训练数据集,将有助于提升性能。因此,模型训练采用了目前最大的蛋白质组数据集——ProteomeTools。借鉴近年来de novo测序领域的进展,研究人员设计了InstaNovo(IN)模型,使用多尺度正弦嵌入编码MS峰值信息,并通过九层Transformer解码器进行预测,同时结合背包束搜索算法确保输出肽段与母离子的m/z一致。
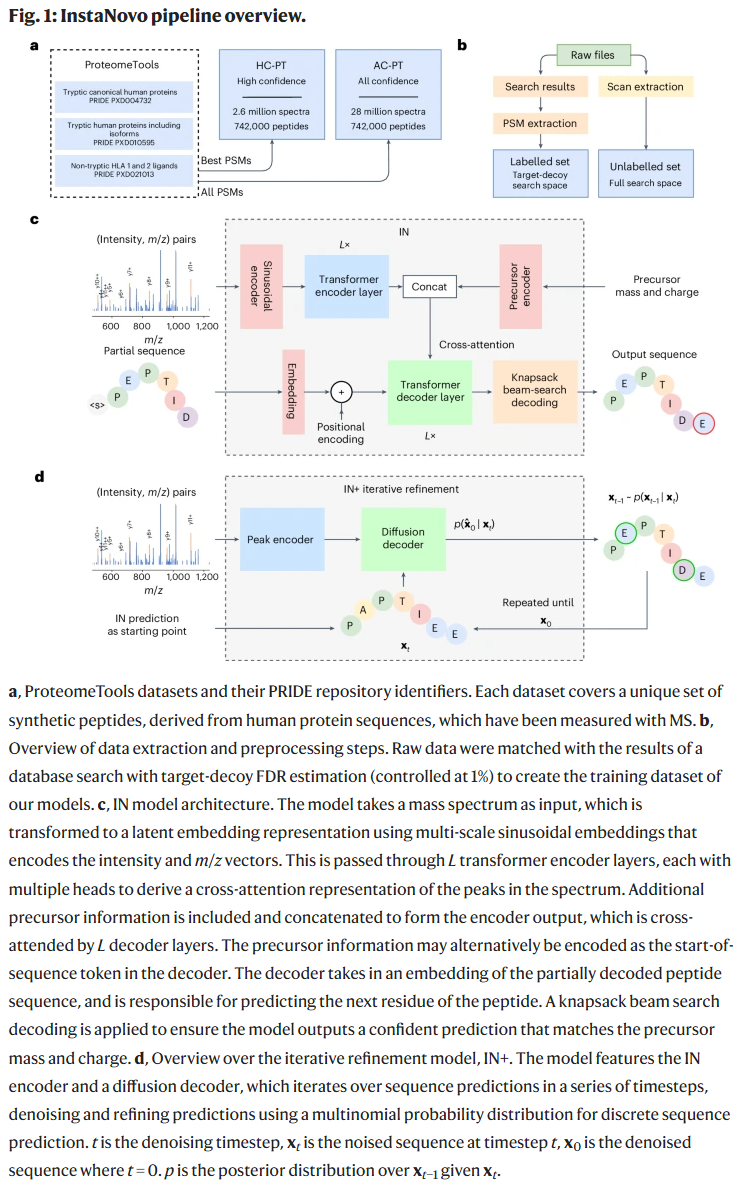
迭代优化提升预测性能
考虑到扩散模型在多个任务中表现优异,研究人员进一步构建了InstaNovo+(IN+)模型,引入迭代精炼机制,模拟人类推断肽段的过程:先给出模糊预测,再逐步修正。该模型包含一个与IN类似的编码器和一个解码器,在20步内不断优化初始预测(可来自随机或IN结果),提升最终准确性。
性能评估
研究人员将IN与当前最强的Transformer模型Casanovo进行了对比,在高分辨九物种数据集和ProteomeTools数据集上评估性能。结果表明,IN和IN+在未经微调时已优于Casanovo,经过微调后IN+表现最佳。在yeast数据集中,IN+以5% FDR预测出52,633个PSMs,较Casanovo多出32.7%;IN与IN+合并后总共预测出56,230个PSMs,提升达41.8%。该优势在其他数据集中亦保持一致。
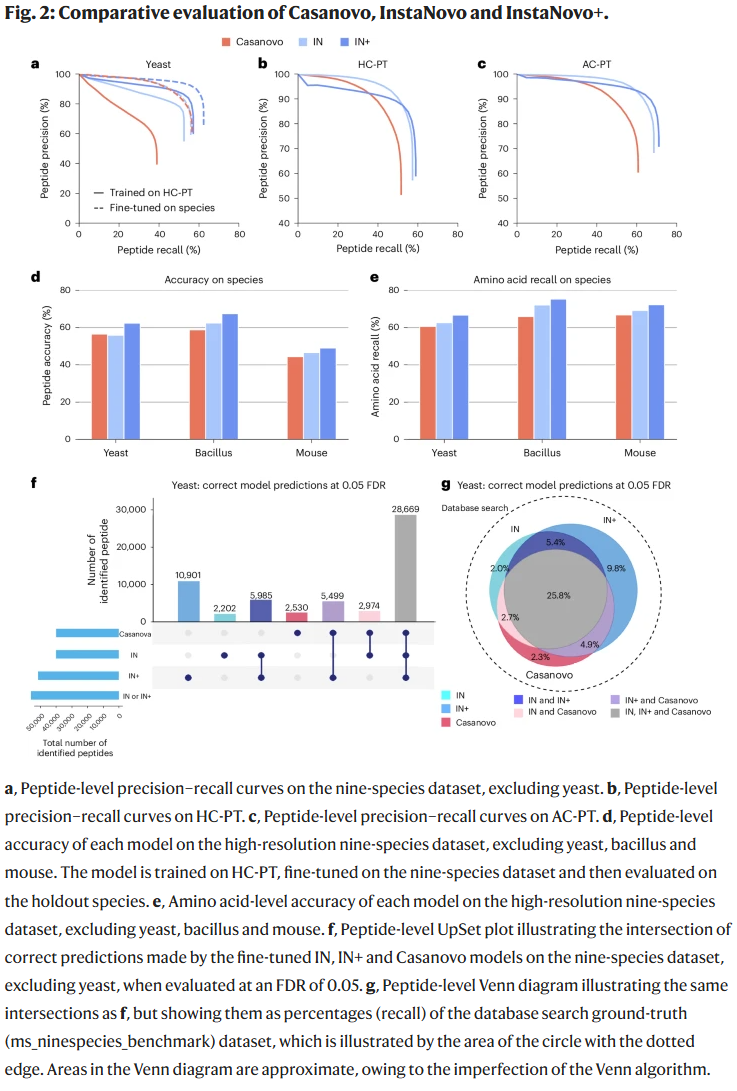
多场景应用评估
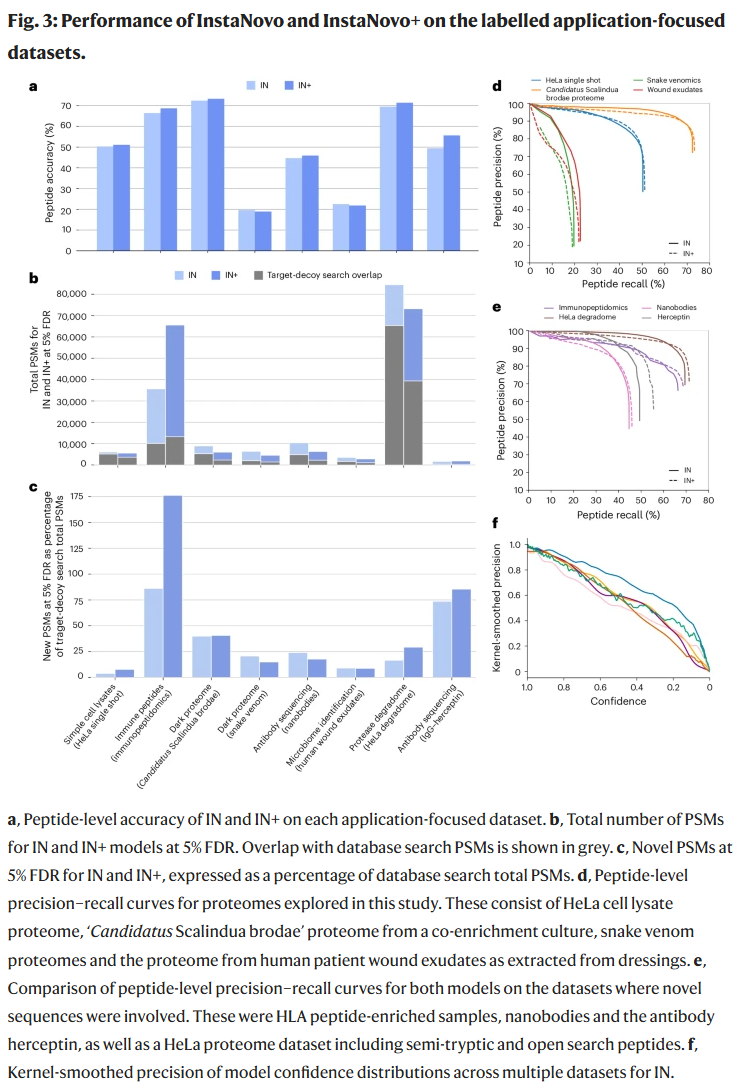
研究人员在8个实际生物数据集上测试了IN与IN+,涵盖单细胞裂解物、免疫肽组、暗蛋白组、抗体测序、微生物组检测等场景。在未进行额外微调的情况下,IN+在Candidatus Scalindua brodae中获得最高73.6%的肽段准确率。平均来看,IN在8个数据集上的肽段准确率为48.3%,IN+为51.5%。在5% FDR下,IN预测的PSMs中约有34%为数据库未识别的新肽段,而IN+在此基础上进一步提升了1.5%的准确率,并发现额外3%的新PSMs。不同样本来源的准确率有所波动,但在高置信区间(>95%)时模型表现总体稳定。
InstaNovo可识别HeLa来源人类蛋白组的过半内容,并提升新型生物分子的序列覆盖率
研究人员首先在HeLa细胞裂解液上进行了基准测试。结果显示,InstaNovo(IN)在该高度注释的蛋白质组中依然能扩展数据库搜索结果,获得49.6%的召回率,共识别出8,774个与数据库一致的PSMs。在5% FDR阈值下,IN在未匹配的MS/MS谱图中额外识别出1,338个PSMs,使识别率提升7.5%。
随后,研究人员评估了IN在新型合成生物分子上的表现。在纳米抗体的de novo测序中,IN识别出7,536个匹配序列,涵盖613条肽段,相较数据库搜索的PSM数量提高了6倍。每个纳米抗体的独特肽段数量由5条提升至40条,增长了8倍。
研究人员还将模型应用于公开的抗体药物Herceptin质谱测序数据,在不同酶解和碎裂条件下,IN实现了重链92.87%、轻链100%的蛋白覆盖率,并简化了测序流程。
InstaNovo可在复杂蛋白质组中发现新蛋白及病原体
研究人员将IN用于来自静脉性腿溃疡患者伤口渗出液的样本。在此复杂背景中,IN将白蛋白的PSMs扩展至1,225个(包含254条独特肽段),是数据库搜索结果的10倍。进一步分析发现,IN还识别出来自铜绿假单胞菌、大肠杆菌和Citrobacter属**的蛋白序列,并通过16S rRNA基因PCR验证了其中两种病原体的存在。
在宏蛋白质组学应用中,研究人员在“Candidatus Scalindua brodae”的共培养体系中检测到1,937条未匹配至任何数据库的肽段,经比对发现这些序列可能来自Phototrophicales、Scalindua arabica、Phycisphaerales、Bacteroidota、Gemmatimonadota等未预设物种。
此外,IN还应用于亚撒哈拉地区26种毒蛇的蛇毒蛋白组样本,发现多个未被数据库覆盖的新肽段,如源自未输入种类“Naja kaouthia”的MTLP-2同源肽段,提示IN在未知或基因组信息有限的样本中具有重要发现潜力。
InstaNovo可应用于免疫肽组和蛋白酶降解组的肽段识别
研究人员在HLA免疫肽组数据中测试了模型表现。IN相比传统数据库搜索,新增了3,495条肽段,提升了41.53%的识别率;IN+在5% FDR下额外识别了11,392个PSMs,并预测出12,965条新肽段。IN识别出的9-mer肽段具有典型的MHC结合位点偏好,增强了预测可信度。
在蛋白酶降解组方面,研究人员在HeLa细胞蛋白与GluC蛋白酶处理后的样本中应用模型,识别出4,635条新肽段,识别率提高11.29%。其中1,222条新序列符合GluC特异性(切割位点前为谷氨酸)。进一步的靶向质谱验证显示这些肽段的确为高置信度的降解产物,明确表明IN可广泛用于全系统层面蛋白酶底物的识别。
讨论
通过拓展蛋白质组学的应用范围并揭示以往难以触及的蛋白质空间,de novo肽段测序为研究复杂生物系统提供了新路径。研究人员提出了InstaNovo(IN)与InstaNovo+(IN+)模型,并在多个应用场景中评估了其性能,包括工程生物分子的测序、免疫肽组学以及暗蛋白组的探索。相比现有工具如Casanovo,两种模型在肽段识别效率和计算成本方面均表现出显著优势,表明其在bottom-up蛋白质组学中具备取代或补充数据库搜索的潜力。
在总体性能提升的基础上,研究人员进一步展示了模型在多个生物学问题中的实际应用。例如,在HeLa细胞中识别出数据库未能检出的蛋白质、将免疫肽组数据中的肽段数量提升175%、并发现新的蛋白酶切位点。基于模型在8个数据集中的表现,研究人员预期其可广泛适用于不同物种和样本类型,并具备良好的泛化能力。未来,该模型有望在蛋白质基因组学、肠道微生物组以及未注释蛋白形式的探索等研究领域中发挥作用,亦可拓展至单细胞蛋白质组学,提高极低样本量条件下的检测灵敏度。
研究人员认为,针对特定任务(如大规模数据或特定PTM)的微调将增强模型识别天然或诱导的化学修饰能力,拓展其在化学蛋白质组学、翻译后修饰检测及多重定量蛋白质组学中的应用。IN与IN+模型亦可能适用于低分辨率质谱数据及多种碎裂方式,但仍需进一步研究其在不同质谱仪(如TOF或离子阱)以及不同MS/MS扫描分辨率下的表现及置信度。
随着**数据非依赖采集(DIA)**等策略的普及,模型输入结构可进一步适应生成的伪MS2谱图,从而实现更高灵敏度的检测。研究人员也预测,多方法融合搜索、后处理打分算法及集成模型将有助于最大化谱图利用效率,并提高识别率。
值得注意的是,本研究以数据库搜索PSM作为肽段识别的“真值”,但数据库搜索本身亦可能存在错误或遗漏。研究人员认为,IN与IN+可用于验证、修正甚至替代部分数据库识别结果,提高检测灵敏度及预测精度。进一步的多特征打分与谱图相似性过滤可作为预测后处理流程,并为IN+提供更优化的初始输入,以释放扩散模型的迭代精炼潜力。此外,模型在非酶切肽段预测方面表现良好,若结合多种蛋白酶信息进行微调,或将进一步提升蛋白覆盖率和序列解析能力。
总体而言,研究人员指出,与其他Transformer架构应用领域类似,数据规模仍是决定de novo测序模型性能的核心因素。未来可借助公共质谱数据库中的海量数据进一步提升模型表现,并推动模型在微调、蛋白组装与新应用开发方面的广泛应用。
整理 | WJM
参考资料
Eloff, K., Kalogeropoulos, K., Mabona, A. et al. InstaNovo enables diffusion-powered de novo peptide sequencing in large-scale proteomics experiments. Nat Mach Intell (2025).
https://doi.org/10.1038/s42256-025-01019-5
内容中包含的图片若涉及版权问题,请及时与我们联系删除
