
DRUGAI
今天为大家介绍的是来自南方科技大学的钟龙华团队的一篇论文。生物大分子的结构对于药物开发和生物催化至关重要。量子细化(Quantum Refinement, QR)方法在晶体学细化中采用了可靠的量子力学(Quantum Mechanics, QM)方法,显示出提高结构质量甚至修正生物大分子结构的潜力。然而,巨大的计算成本和复杂的量子力学/分子力学(QM/MM)设置限制了QR方法的应用。本文中,作者将稳健的机器学习势能(Machine Learning Potentials, MLPs)引入到多尺度ONIOM(QM:MM)方案中用以描述核心部分(如药物/抑制剂),替代昂贵的QM方法。此外,作者首次结合了两个层级的MLPs以克服MLPs的局限性。作者独特的基于MLPs+ONIOM的QR方法在实现QM级别精度的同时,具有显著更高的效率。此外,细化结果提供了FDA批准的药物nirmatrelvir在SARS-CoV-2主要蛋白酶结构中存在结合和非结合形式的计算证据。本研究强调了强大的MLPs加速了可靠的蛋白质-药物复合物的QR,促进了更广泛的QR应用,并为药物开发提供了更多原子级的见解。

生物大分子的精确原子结构对于分子性质预测、结合位点估计以及理解配体结合位点识别和生物催化至关重要。这些结构信息在高效且专一性强的新药物开发和设计中起着不可或缺的作用。X射线衍射(XRD)是确定许多生物大分子原子结构的强大方法,常结合分子力学(MM)力场和实验数据。然而,开发可靠的力场以涵盖多样化的化学空间和复杂电子效应一直是个挑战。尽管AI方法(如AlphaFold)在预测蛋白质结构方面取得进展,但对于包含辅因子或药物/抑制剂的生物结构预测仍有限。量子细化(QR)方法通过更精确的量子力学(QM)替代MM方法,克服了这些挑战,并已成功应用于多个蛋白质-药物系统。QR方法结合多尺度、线性尺度QM、碎片化和量子嵌入等方法,进一步提升了细化过程。
然而,与快速的MM方法相比,QM/MM系统的高计算成本和复杂设置限制了QR在生物系统中的广泛应用。最近,机器学习势能(MLPs)作为一种有前景的替代方案,能够快速预测能量和梯度,展示出与高水平从头计算方法相当的精度。
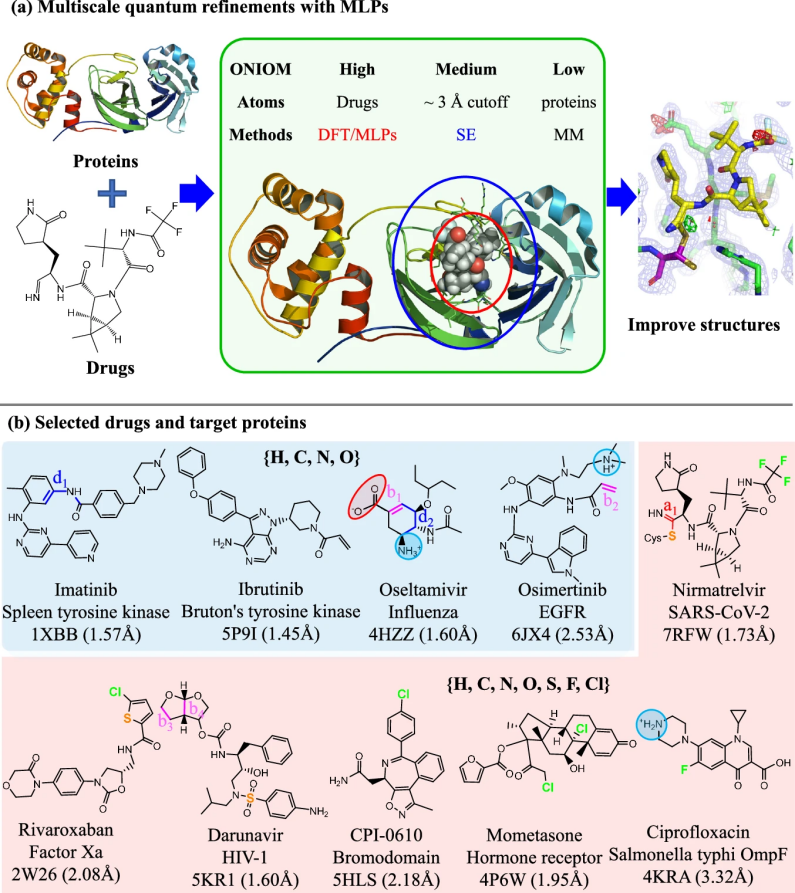
图1:工作流程示意图及选择的蛋白质-药物/抑制剂系统
在本研究中,如图1a所示,机器学习势能(MLPs)首次作为多尺度量子细化(QR)方法中的高层引入,其中昂贵的量子力学(QM)方法被更快的ANI或AIQM1方法取代。由于对训练数据的高度依赖,MLPs仅限于少数元素(例如AIQM1:C、H、O、N;ANI-2x:C、H、O、N、F、Cl、S)或特定系统(ANI:中性系统)。为了对包含更多元素的药物/抑制剂分子进行细化,并在保持核心药物/抑制剂结构的最高精度的同时克服这种限制,作者首次通过外推的N层集成分子轨道和分子力学(ONIOM)方法结合了两个不同水平(CC和DFT级别)的MLPs(分别称为MLP-CC和MLP-DFT),引入ONIOM QR方案中。
气相中的药物/抑制剂结构
表2:不同数据集下气相优化结构的比较
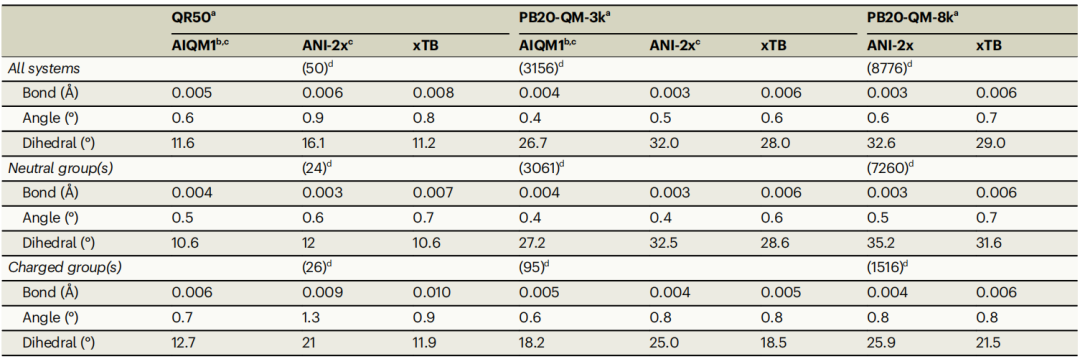
如图1b所示,为了评估一些MLPs的性能,首先对选择的50种药物/抑制剂在气相中使用ωB97X-D/6-31G(d)、AIQM1、ANI-1ccx、ANI-2x、ANI-1x和第二代几何、频率、非共价、扩展紧密结合(GFN2-xTB)方法进行几何优化。与(QM)ωB97X-D方法优化的可靠结果相比,MLPs(AIQM1、ANI-2x)和SE(GFN2-xTB)方法在键长、角度和可旋转二面角上的中位绝对偏差(MAD)分别为0.005–0.008Å、0.6–0.9°和11.2–16.1°(表2)。
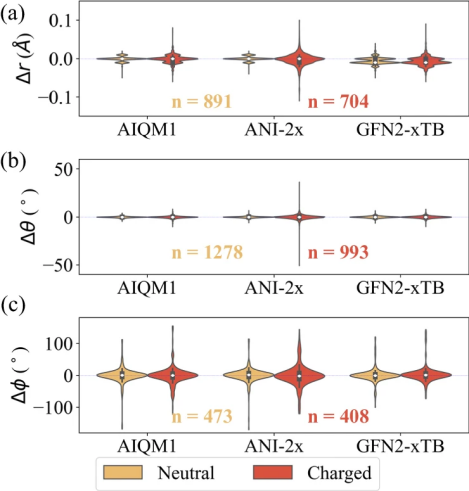
图2:气相中优化结构比较
总体而言,所有MLPs导致的药物/抑制剂结构与DFT方法优化的结构相似,所有中位数(白点)和最大偏差分布(最大宽度)位置接近零(图2)。然而,GFN2-xTB方法低估了键长,中位键偏差分布接近-0.01Å。
蛋白质-药物/抑制剂系统的量子细化
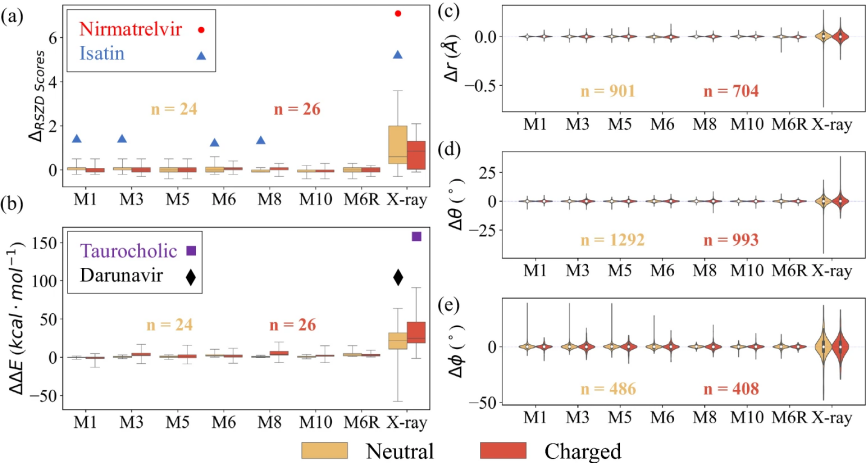
图3:量子细化结果的比较
这些MLPs进一步被引入基于ONIOM的QR方案中,以细化50个蛋白质-药物/抑制剂系统的结构。与X射线晶体结构相比,这50种药物/抑制剂在各种QR方法后的实空间差异密度Z(RSZD)得分平均减少了1.0–1.1(图3a),表明结构经过QR方法有了改进。最显著的改进出现在SARS-CoV-2 Mpro系统中的nirmatrelvir和DJ-1系统中的isatin,其RSZD得分由于考虑了两种构象分别从7.6/6.0(X射线晶体结构)显著降低到0.5–1.0/0.9–2.1。
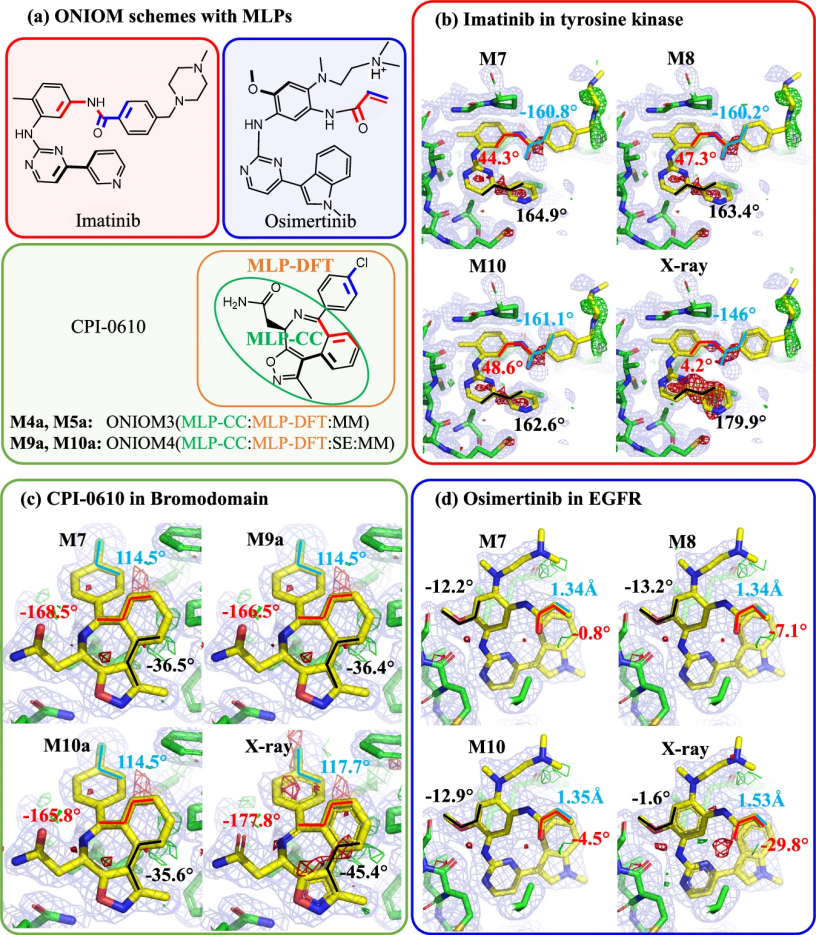
图4:结合MLPs的ONIOM方法以及电子密度图
如图4所示,与上述QR方法后RSZD得分的减少一致,电子密度分析也显示出所有细化的药物/抑制剂结合位点周围的电子密度显著改善。例如,imatinib、osimertinib和CPI-0610系统的电子密度图在作者的QR方法后显示出更好的密度拟合。此外,与X射线电子密度图相比,实验观测结果的差异显著减少(即更少的绿色和/或红色轮廓)。
此外,这50种药物/抑制剂的计算应变能(图3b)在各种QR方法后平均减少了27.1~31.4 kcal·mol−1。应变能减少最大的是taurocholic acid-CmeR系统(从239.8减少到约78.7–85.6 kcal·mol−1)和darunavir-HIV-1系统(从117.3减少到约14.1–20.4 kcal·mol−1)。这种应变能减少可以归因于X射线结构中低估的taurocholic acid的C–S键(b5:减短了0.18–0.22 Å)和darunavir的两个C–C键(b3和b4:减短了0.25~0.28 Å)因此,这些计算结果表明,与X射线晶体结构相比,所有QR方案(M1–M10)都显著改进了局部药物/抑制剂结合位点。
此外,结构分析(图3c–e)进一步揭示了各种QR方案细化后的药物/抑制剂的键长、角度和可旋转二面角与最可靠的ONIOM3(DFT:SE:MM)方法(M7)保持一致。与X射线结构相比,使用方法M1–M10细化的darunavir、oseltamivir和osimertinib结构显示了最大的绝对键偏差,这可以解释这些X射线结构中计算出的非常高的应变能。
通过ONIOM方法结合两种MLPs
由于CC级别的MLPs(ANI-1ccx和AIQM1)只能应用于有限的元素(H、C、N、O),这些高级别的MLPs无法用于描述包含更多元素(F、S、Cl)的更广泛的药物/抑制剂系统。为了克服这一限制并描述更多的药物/抑制剂系统,作者首次通过外推的ONIOM方案结合了两种不同水平的MLPs,其中主要核心结构由高级别MLP-CC方法(ANI-1ccx或AIQM1)描述,包含其他元素的部分由低级别MLP-DFT(ANI-2x)方法描述(图4a)。这种新的两种MLPs的组合能够在20个包含F、Cl或S元素的选择蛋白质-药物/抑制剂系统上进行前所未有的ONIOM3和ONIOM4方案。
如图4c所示,通过这些基于ONIOM4的方案得到的CPI-0610在溴结构域中的细化结构在结合位点周围的电子密度与可靠的ONIOM3(DFT:SE:MM)M7方案相似,与实验观测结果的差异减少(即更少的红色和/或绿色轮廓)。这些发现表明,这些独特的ONIOM4(MLP-CC:MLP-DFT:SE:MM)和ONIOM3(MLP-CC:MLP-DFT:MM)方案可以以更高的计算效率改善蛋白质-药物/抑制剂结构,表明通过ONIOM方法结合多个层级的MLPs为克服MLPs的一些限制并增强其优势提供了一条有前景的途径。
相关性
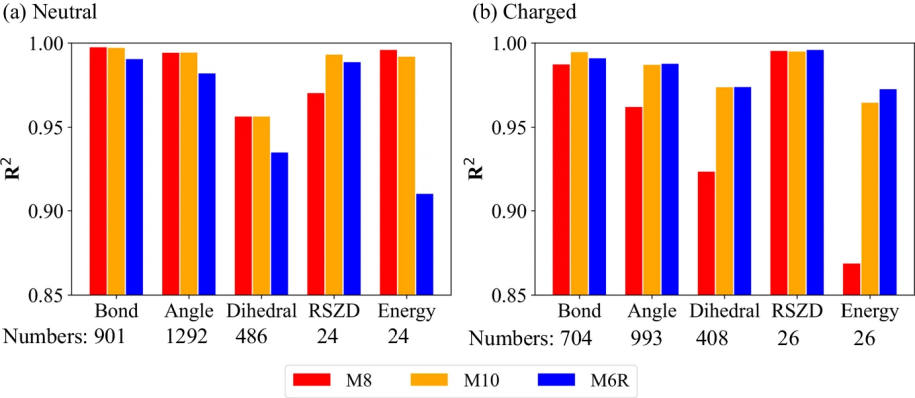
图5:不同QR方案的准确性
图5显示了通过M8、M10和M6R方案(分别为ONIOM3(ANI-2x:SE:MM)、ONIOM3(AIQM1:SE:MM)和ONIOM2(SE:MM))获得的细化键长、角度、可旋转二面角、RSZD得分和应变能与通过M7方案获得的结果的相关性(R²)。通过M8、M10和M6R获得的细化键长和角度与通过M7方案获得的结果非常一致(键长:R²>0.988;角度:R²>0.962),适用于包含带电和中性基团的药物/抑制剂系统。相反,它们的可旋转二面角、RSZD得分和应变能在包含中性和带电基团的系统之间显示出明显差异。
气相和蛋白质中的结构比较

图6:在气相和蛋白质中的结构比较
相对于DFT方法,图6展示了所有计算的药物/抑制剂结构在气相和蛋白质中的RMSDs。在气相中优化的药物/抑制剂结构的RMSDs显著高于通过QR细化的蛋白质中的相应结构。这些结果可以归因于气相中结构的高度灵活性,而蛋白质结合位点的空间受限程度较高。此外,当使用ANI-2x方法时,含有带电基团的药物/抑制剂的RMSDs略大于中性药物/抑制剂,这可能是由于前述ANI方法的限制。高级别的AIQM1方法在气相中优化的药物/抑制剂结构中表现更好(带电:0.49 Å;中性:0.59 Å),优于ANI-2x方法(带电:0.74 Å;中性:0.67 Å)。这些结果表明,尽管在气相中观察到较大的结构误差,但使用MLPs(特别是更精确的CC级别的AIQM1)进行的QR方法可以在蛋白质中提供可靠的药物/抑制剂结构。
SARS-CoV-2 MPro中nirmatrelvir的两种构象
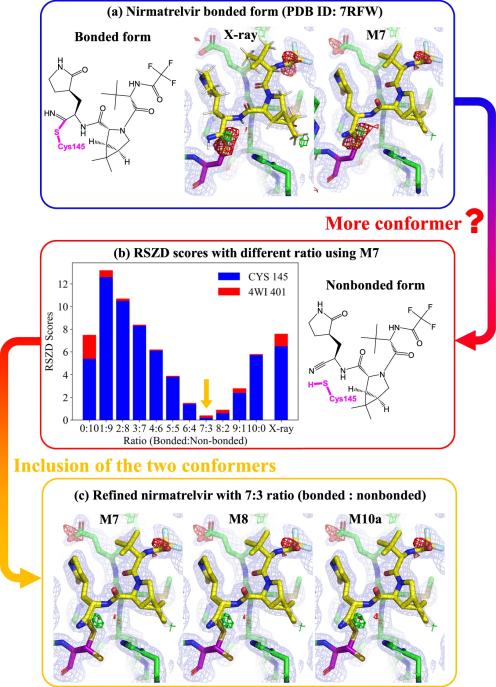
图7:SARS-CoV-2 MPro的量子细化
如图7a所示,在野生型SARS-CoV-2 MPro晶体结构中,nirmatrelvir结合位点周围的电子密度图显示在与Cys145的C–S共价键连接处有明显的红色轮廓。然而,通过几种方案(M1-M10)进行的QR仅略微减轻了这一电子密度的显著差异,并将RSZD得分从7.6略微降低到5.8。基于可逆的C–S键形成机制,作者的QR方法进一步考虑了结合和非结合构象的存在可能性。
作者使用基于DFT的ONIOM3(DFT:SE:MM)M7方案对这两种构象分别进行细化,然后将细化结果与两种构象的不同占比相结合。细化结果表明,约7:3(结合:非结合)的占比给出了最大的结构改进和最低的RSZD得分(0.5,图7b),以及显著改进的电子密度(图7c)。使用基于MLP的方案(M8-M10)细化的结果与使用基于DFT的方案(M7)细化的结果非常相似,RSZD得分仅相差
讨论
在本研究中,作者首次提出并使用机器学习势能替代可靠但昂贵的量子力学方法,以加速多尺度量子细化过程。为克服一些MLPs的元素限制,采用外推ONIOM方法结合了两个不同级别的MLPs,并应用于前所未有的ONIOM3和ONIOM4方案进行QR。概念验证研究表明,强大的MLPs可以以高精度加速蛋白质-药物/抑制剂复合物的QR过程,这将促进更多的QR应用,并为分子识别、催化和药物开发提供新的原子级见解。此外,除了X射线晶体学,作者认为MLPs还可以对现代生物大分子结构测定方法有帮助。作者还建立了计算基准数据集(PB20-QM、PB20-QM-8k和PB20-QM-3k),以评估药物/抑制剂分子的结构可靠性,希望这能帮助未来开发更好的用于药物/抑制剂分子的DFT、MLPs或SE方法。作者也乐观地认为,随着不同研究组开发的更多高级和通用MLPs的进展,未来将在普通台式电脑上常规进行各种生物系统的快速可靠的QR。
编译 | 于洲
审稿 | 曾全晨
参考资料
Yan Z, Wei D, Li X, et al. Accelerating reliable multiscale quantum refinement of protein–drug systems enabled by machine learning[J]. Nature Communications, 2024, 15(1): 4181.
