

人类基因组中的结构变异(SV)与遗传多样性和许多常见、罕见疾病有关。由于SV的复杂性和测序技术的固有局限性,一直以来,SV检测都是基因组研究领域的国际性难题。2023年,人类泛基因组参考联盟(HPRC)发布了由多个基因组测序平台生成的44个二倍体长读长测序序列的泛基因组草案,并揭示了这种参考图谱如何增强SV的发现。但目前具有全球代表性的人口规模的长读长测序SV数据集仍然有限。
近日,在Nature发表的一项最新研究中,欧洲分子生物学实验室(EMBL)、欧洲生物信息学研究所(EBI)、德国杜塞尔多夫海因里希·海涅大学等团队合作利用长读长测序(ONT)分析了来自千人基因组计划(1kGP)样本的SV。通过整合线性和基于图谱的基因组分析,研究发现了超过10万个序列分辨的双等位基因SV,并对30万个多等位基因可变串联重复序列(VNTR)进行了基因分型。该研究生成的泛基因组数据集构成了一个全面的DNA序列解析的SV等位基因图谱,涵盖了从常见到罕见的变异。该数据集提供了一个使用长读长测序技术的全球遗传变异参考,促进了SV生物学和人类疾病研究,并对SV的等位基因结构、机制起源、突变复发和人群分布提供了新见解。

文章共同通讯作者、EMBL临时负责人Jan Korbel表示:“利用新的测序技术对人类基因组进行常规分析已经成为可能,这些技术可以解码更长的DNA片段,使我们能够组装个人的完整基因组,并评估基因组的SV等所有遗传变异。我们可以利用这些新的变革性测序技术的力量来更多地了解人类遗传变异。”
文章共同通讯作者、德国杜塞尔多夫海因里希·海涅大学实验室Tobias Marschall博士表示:“该研究为人类的SV等遗传变异提供了参考,尤其有助于罕见病的研究,因为它能让我们区分良性变异和可能导致疾病的变异。”
为了包含广泛的单倍型多样性,研究团队对来自五大地区(欧洲、东亚、南亚、非洲、美洲)26个祖先群体1019人的样本进行了长读长测序,测序的中位覆盖率为16.9x,中位N50读长为20.3kb。为了进行图形感知的SV发现和基因分型,研究团队设计了SAGA(SV analysis by graph augmentation)算法(图1c)。
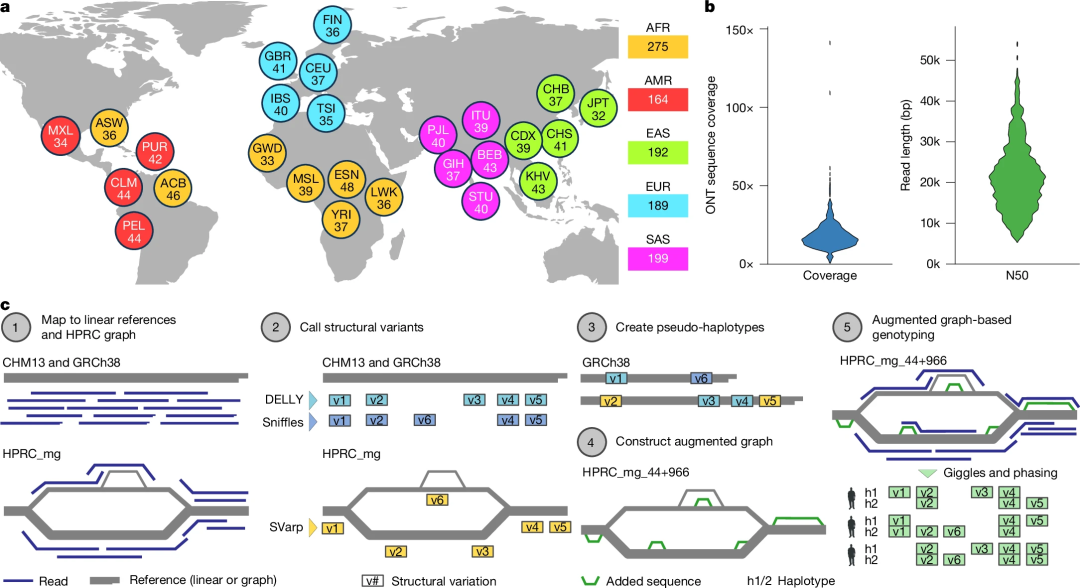
图1.1019名个体的长读长测序和SAGA算法。
通过整合线性和基于图谱的分析,研究团队在每一份样本中挖掘出15,301~21,529个SV,并在967份长读长测序样本的子集中识别出167,291个初级SV位点。进一步SV基因分型和分相分析显示,164,571个(98.4%)已测序的SV位点成功进行了分相。这些构成了最终的基于SAGA的SV调用集,包括65,075个缺失、74,125个插入和25,371个“假定为复杂”的位点,这些位点的参考等位基因和替代等位基因都大于1bp。该SV调用集中107,005个为双等位基因SV,其余为多等位基因SV。
研究团队分析了在每次添加新基因组后,该SV调用数据集的变化,发现常见SV存在明显的饱和效应,非洲裔样本的SV数量不成比例地增加。与之前在1kGP样本中进行的群体规模基因组测序研究相比,该数据集的SV谱中捕获了更多的变异,SV范围从50~几百bp,这使用短读长测序很难捕获。
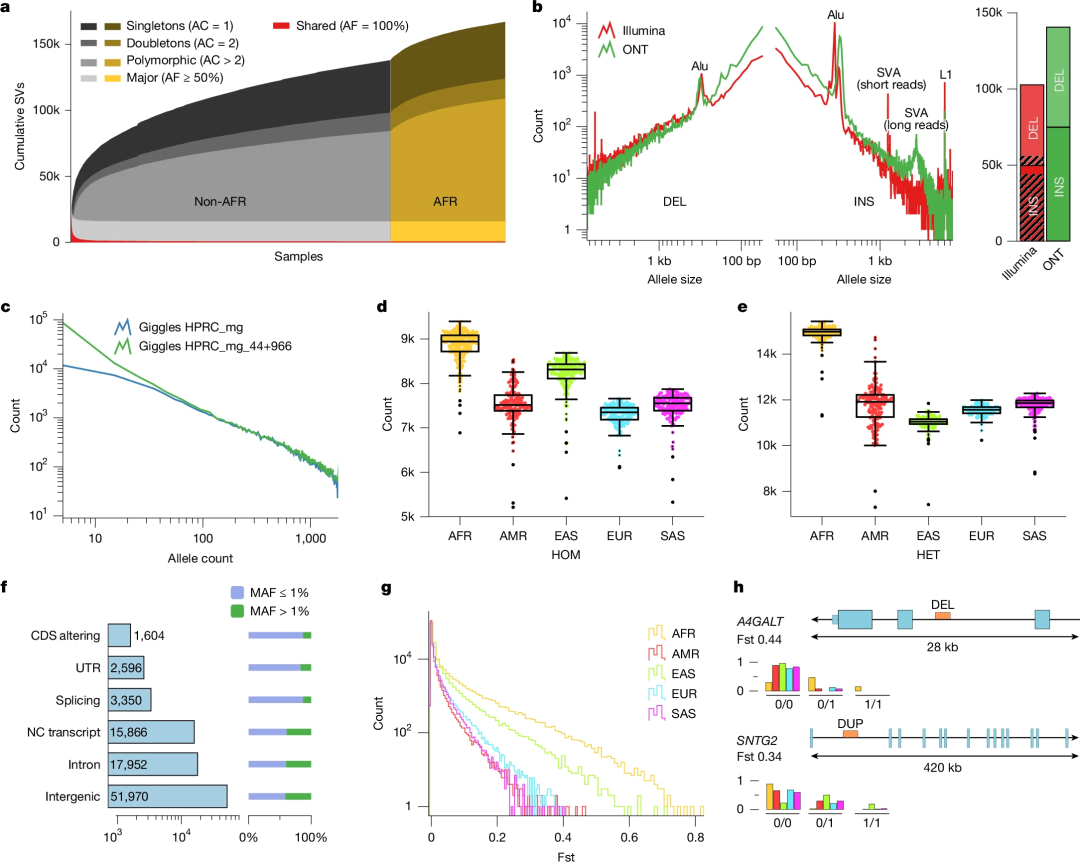
图2.26个祖先人群的SV图谱。
数据显示,非洲裔样本的每个样本SV中位数为23,969,非非洲裔为19,297。相比之下,对相同样本进行子集划分时,通过短读长测序检测到的非洲裔和非非洲裔SV中位数分别为9,963和8,540。其中获得最大增益的是插入,该数据集能以更高的灵敏度捕获插入,并且SV等位基因的插入位点增加了十倍以上(图2b)。对于缺失,该数据集将SV数量增加了40%(从46,895增加到65,812)。
该数据集中大多数SV是罕见的(59.3%的MAF)。在当前样本量下,多数SV通常在单一队列中被检测到(非洲、美洲、亚洲东部、欧洲或撒哈拉以南非洲)。相比之下,等位基因频率≥2.5%的大多数SV至少出现在两个地区群体中。多等位基因SV位点通常比双等位基因SV更倾向于跨地区共享,大多数等位基因频率≥1.5%的多等位基因SV是共享的。此外,非洲样本中杂合子SV增加高于纯合子,这反映了非洲样本更大的遗传多样性。
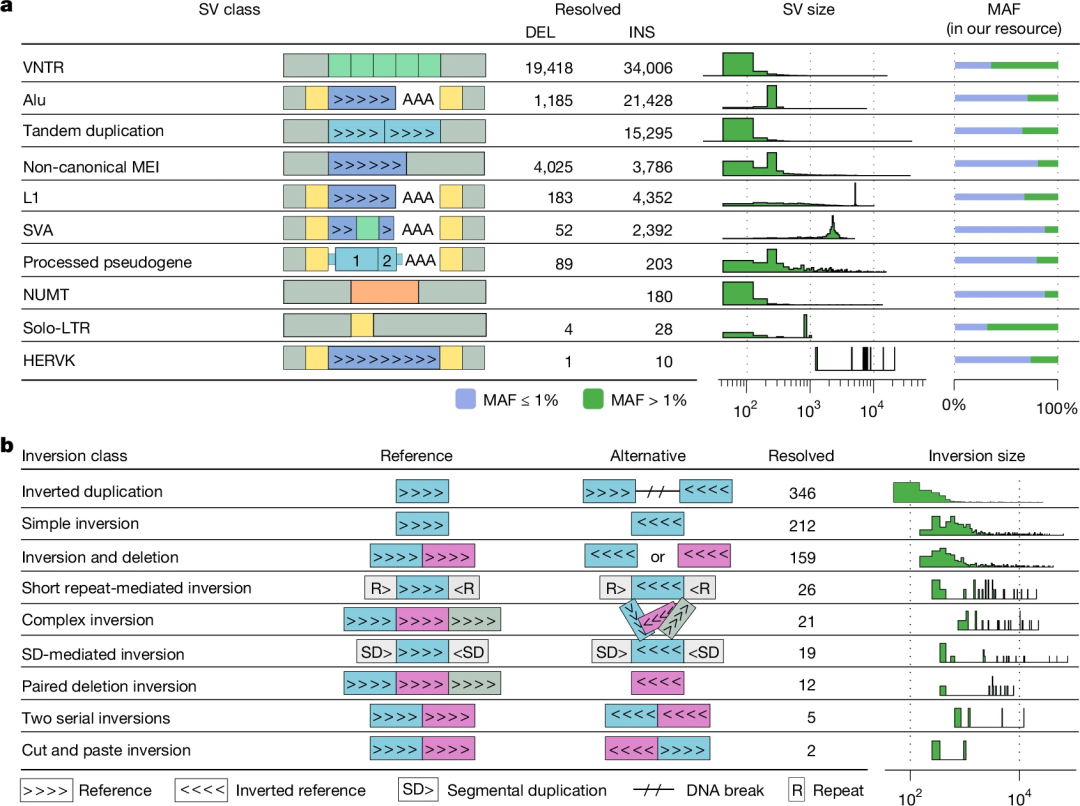
图3.基于SAGA的数据集中不同SV类型的流行度。
该数据集中,有17,029个(19.5%)插入是重复序列,其中15,295个(89.8%)为串联重复。此外,有34,006个(39.0%)插入和19,418个(26.2%)缺失被归类为VNTR,并且大多数VNTR(50.4%)代表多等位基因SV位点。
对于倒位,研究发现在显示成簇错配的区域进行reads重新比对可以提高倒位检测的准确性,共发现1849个倒位调用。同时,研究人员利用该数据集分析了所有序列解析的多态性移动元件(MEI)之间的多态性转导事件,注释了878个转导事件,其中5,311个L1插入中有466个(8.8%),3,470个SVA插入中有412个(11.9%)表现出转导现象。此外,研究发现TEMR对人类基因组中复发SV形成有重要贡献。
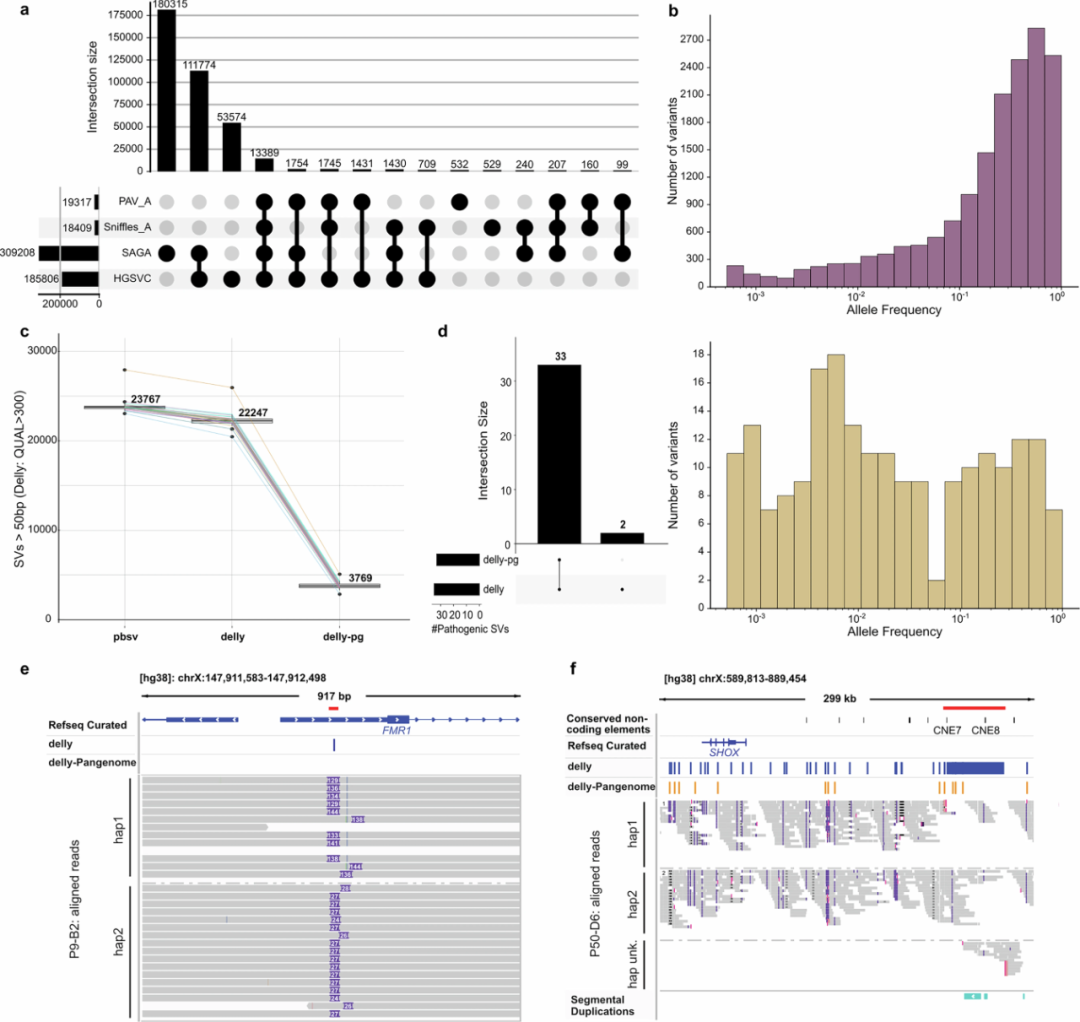
图4.使用SVAN注释的MEI序列特征。
此外,研究团队深入挖掘了四名被诊断患有罕见病个体的长读长测序数据,平均每个患者发现了386个候选SV,并分析了大约270个疾病相关基因,这些基因位于基因组复杂的部分,预示着未来疾病研究中可能改进的基因分型。
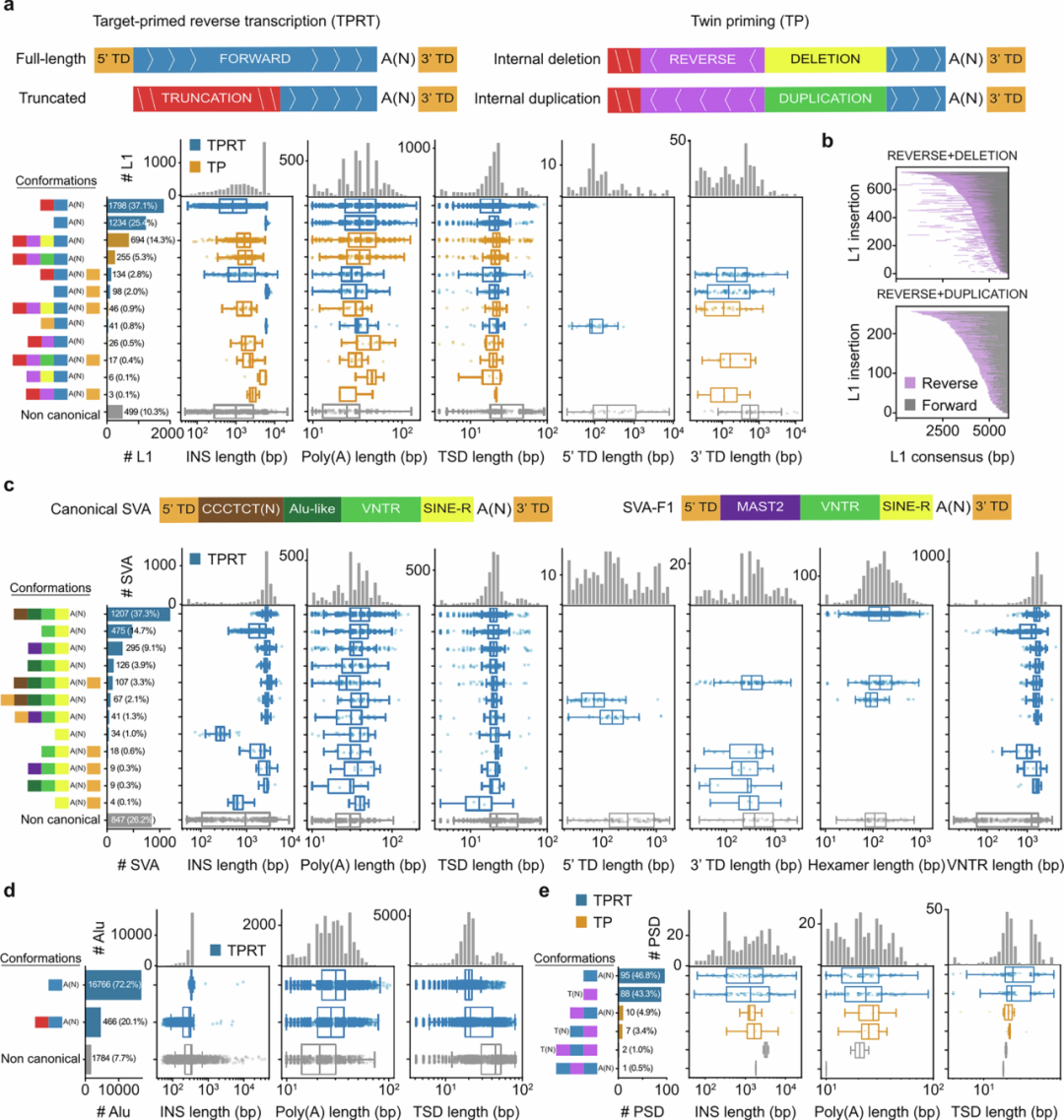
图5.患者基因组分析。
综上,该研究绘制了涵盖26个祖先人群中常见和罕见SV图谱。与gnomAD相比,该图谱数据中有50.9%的插入、14.5%的缺失尚未被报道,强调了长读长测序在推进SV表征方面的价值。
文章作者、EMBL-EBI研究员Sarah Hunt表示:“最初的1kGP计划绘制了人类群体中的基因组变异图谱,帮助我们系统地分析常见疾病相关区域。该研究提供的新资源比迄今为止创建的其他SV图谱更精确、更深入,这将使我们能够寻找新的疾病联系。”
值得注意的是,在Nature同期发表的另一篇文章中,Korbel和Marschall及其合作者利用PacBio HiFi测序和ONT长读长测序等技术,完成了来自20多个不同人类群体65个个体的130个单倍型解析的基因组组装。该研究弥补了此前基因组组装92%缺口,39%的染色体获得端粒到端粒序列,并分析了复杂SV和区域。
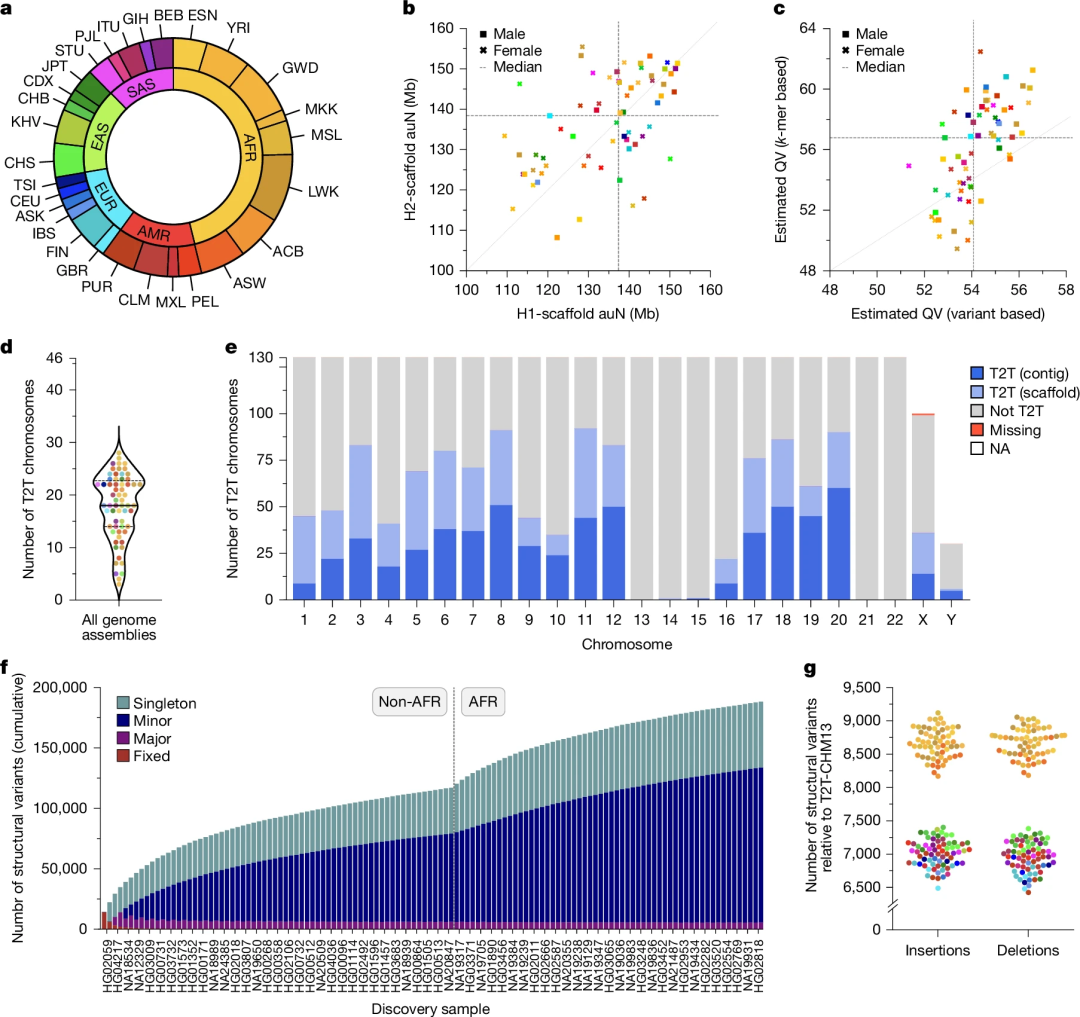
图6.65个不同人类的全基因组测序、组装和变异检测。
此外,EMBL并不是唯一一个对1KGP样本进行长读长测序的团队,由华盛顿大学Danny Miller领导的千人基因组计划长读长测序(1KGP-LRS)联盟,同样使用了这一技术。近日,该项目重新定位并扩展,将对全部1KGP样本的DNA和RNA进行PacBio、ONT测序,期望从数据中挖掘出更多的生物学见解,例如基因调控、选择性剪接和转录组复杂性等。
Miller表示,1KGP-LRS联盟还计划与上述研究团队、HPRC等合作整合数据集,为更广泛的研究提供关于1KGP项目样本更全面的资源。
参考资料:
1.Schloissnig, S., Pani, S., Ebler, J. et al. Structural variation in 1,019 diverse humans based on long-read sequencing. Nature (2025). https://doi.org/10.1038/s41586-025-09290-7
2.Logsdon, G.A., Ebert, P., Audano, P.A. et al. Complex genetic variation in nearly complete human genomes. Nature (2025). https://doi.org/10.1038/s41586-025-09140-6
3.https://www.genomeweb.com/genetic-research/long-read-sequencing-reveals-new-structural-variation-details-1000-genomes-project
4.https://www.genomeweb.com/sequencing/uw-led-1000-genomes-project-long-read-sequencing-consortium-adds-pacbio-data
内容中包含的图片若涉及版权问题,请及时与我们联系删除
