
[This is one of the finalists in the 2025 review contest, written by an ACX reader who will remain anonymous until after voting is done. I’ll be posting about one of these a week for several months. When you’ve read them all, I’ll ask you to vote for a favorite, so remember which ones you liked]
“The scientific paper is a ‘fraud’ that creates “a totally misleading narrative of the processes of thought that go into the making of scientific discoveries.”
This critique comes not from a conspiracist on the margins of science, but from Nobel laureate Sir Peter Medawar. A brilliant experimentalist whose work on immune tolerance laid the foundation for modern organ transplantation, Sir Peter understood both the power and the limitations of scientific communication.
Consider the familiar structure of a scientific paper: Introduction (background and hypothesis), Methods, Results, Discussion, Conclusion. This format implies that the work followed a clean, sequential progression: scientists identified a gap in knowledge, formulated a causal explanation, designed definitive experiments to fill the gap, evaluated compelling results, and most of the time, confirmed their hypothesis.
Real lab work rarely follows such a clear path. Biological research is filled with what Medawar describes lovingly as “messing about”: false starts, starting in the middle, unexpected results, reformulated hypotheses, and intriguing accidental findings. The published paper ignores the mess in favour of the illusion of structure and discipline. It offers an ideal version of what might have happened rather than a confession of what did.
The polish serves a purpose. It makes complex work accessible (at least if you work in the same or a similar field!). It allows researchers to build upon new findings.
But the contrived omissions can also play upon even the most well-regarded scientist’s susceptibility to the seduction of story. As Christophe Bernard, Director of Research at the Institute of Systems Neuroscience (Marseilles, Fr.) recently explained,
“when we are reading a paper, we tend to follow the reasoning and logic of the authors, and if the argumentation is nicely laid out, it is difficult to pause, take a step back, and try to get an overall picture.”
Our minds travel the narrative path laid out for us, making it harder to spot potential flaws in logic or alternative interpretations of the data, and making conclusions feel far more definitive than they often are.
Medawar’s framing is my compass when I do deep dives into major discoveries in translational neuroscience. I approach papers with a dual vision. First, what is actually presented? But second, and often more importantly, what is not shown? How was the work likely done in reality? What alternatives were tried but not reported? What assumptions guided the experimental design? What other interpretations might fit the data if the results are not as convincing or cohesive as argued?
And what are the consequences for scientific progress?
In the case of Alzheimer’s research, they appear to be stark: thirty years of prioritizing an incomplete model of the disease’s causes; billions of corporate, government, and foundation dollars spent pursuing a narrow path to drug development; the relative exclusion of alternative hypotheses from funding opportunities and attention; and little progress toward disease-modifying treatments or a cure.
The incomplete Alzheimer’s model I’m referring to is the amyloid cascade hypothesis, which proposes that Alzheimer’s is the outcome of protein processing gone awry in the brain, leading to the production of plaques that trigger a cascade of other pathological changes, ultimately causing the cognitive decline we recognize as the disease. Amyloid work continues to dominate the research and drug development landscape, giving the hypothesis the aura of settled fact.
However, cracks are showing in this façade. In 2021, the FDA granted accelerated approval to aducanumab (Aduhelm), an anti-amyloid drug developed by Biogen, despite scant evidence that it meaningfully altered the course of cognitive decline. The decision to approve, made over near-unanimous opposition from the agency’s advisory panel, exposed growing tensions between regulatory optimism and scientific rigor. Medicare’s subsequent decision to restrict coverage to clinical trials, and Biogen’s quiet withdrawal of the drug from broader marketing efforts in 2024, made the disconnect impossible to ignore.
Meanwhile, a deeper fissure emerged: an investigation by Science unearthed evidence of data fabrication surrounding research on Aβ*56, a purported toxic amyloid-beta oligomer once hailed as a breakthrough target for disease-modifying therapy. Research results that had been seen as a promising pivot in the evolution of the amyloid cascade hypothesis, a new hope for rescuing the theory after repeated clinical failures, now appears to have been largely a sham. Treating Alzheimer’s by targeting amyloid plaques may have been a null path from the start.
When the cracks run that deep, it’s worth going back to the origin story—a landmark 1995 paper by Games et al., featured on the cover of Nature under the headline “A mouse model for Alzheimer’s.” It announced what was hailed as a breakthrough: the first genetically engineered mouse designed to mimic key features of the disease.
In what follows, I argue that the seeds of today’s failures were visible from the beginning if one looks carefully. I approach this review not as an Alzheimer’s researcher with a rival theory, but as a molecular neuroscientist interested in how fields sometimes converge around alluring but unstable ideas. Foundational papers deserve special scrutiny because they become the bedrock for decades of research. When that bedrock slips beneath us, it tells a cautionary story: about the power of narrative, the comfort of consensus, and the dangers of devotion without durable evidence. It also reminds us that while science is ultimately self-correcting, correction can be glacial when careers and reputations are staked on fragile ground.
The Rise of the Amyloid Hypothesis
In the early 1990s, a new idea began to dominate Alzheimer’s research: the amyloid cascade hypothesis.
First proposed by Hardy and Higgins in a 1992 Science perspective, the hypothesis suggested a clear sequence of disease-precipitating events: protein processing goes awry in the brain → beta-amyloid (Aβ) accumulates → plaques form → plaques trigger a cascade of downstream events, including neurofibrillary tangles, inflammation, synaptic loss, neuronal death, resulting in observable cognitive decline.
The hypothesis was compelling for several reasons. First, the discovery of the enzymatic steps by which amyloid precursor protein (APP) is processed into Aβ offered multiple potential intervention points—ideal for pharmaceutical drug development.
Second, the hypothesis was backed by powerful genetic evidence. Mutations in the APP gene on chromosome 21 were associated with early-onset Alzheimer’s. The case grew stronger with the observation that more than 50% of individuals with Down syndrome, who carry an extra copy of chromosome 21 (and thus extra APP), develop Alzheimer’s-like pathology by age 40.
Thus, like any robust causal theory, the amyloid cascade hypothesis offered explicit, testable predictions. As Hardy and Higgins outlined, if amyloid truly initiates the Alzheimer’s cascade, then genetically engineering mice to produce human amyloid should trigger the full sequence of events: plaques first, then tangles, synapse loss, and neuronal death, then cognitive decline. And the sequentiality matters: amyloid accumulation should precede other pathological features. At the time, this was a thrilling possibility.
Pharmaceutical companies were especially eager: if the hypothesis proved correct, stopping amyloid should stop the disease. The field awaited the first transgenic mouse studies with enormous anticipation.
How—with Unlimited Time and Money and a Little Scientific Despair—to Make a Transgenic Mouse
“Mouse Model Made” was the boastful headline to the independent, introductory commentary Nature solicited to accompany the 1995 Games paper’s unveiling of the first transgenic mouse set to “answer the needs” of Alzheimer’s research. The scientific argument over whether amyloid caused Alzheimer’s had been “settle[d]” by the Games paper, “perhaps for good.”
In some ways, the commentary’s bravado seemed warranted. Why? Because in the mid-’90s, creating a transgenic mouse was a multi-stage, treacherous gauntlet of molecular biology. Every step carried an uncomfortably high chance of failure. If this mouse, developed by Athena Neurosciences (a small Bay Area pharmaceutical company) was valid, it was an extraordinary technical achievement portending a revolution in Alzheimer’s care.
First Rule of Making a Transgenic Mouse: Don’t Talk About How You Made a Transgenic Mouse
How did Athena pull it off? Hard to say! What's most remarkable about the Games paper is what's not there. Scan through the methods section and you'll find virtually none of the painstaking effort required to build the Alzheimer’s mouse. Back in the ‘90s, creating a transgenic mouse took years of work, countless failed attempts, and extraordinary technical skill. In the Games paper, this effort is compressed into a few sparse sentences describing which gene and promoter (nearby gene instruction code) the research team used to make the mouse. The actual details are relegated to scientific meta-narrative—knowledge that exists only in lab notebooks, daily conversations between scientists, and the muscle memory of researchers who perform these techniques thousands of times.
The thin description wasn’t atypical for a publication from this era. Difficult experimental methods were often encapsulated in the single phrase "steps were carried out according to standard procedures," with citations to entire books on sub-cloning techniques or reference to the venerable Manipulating the Mouse Embryo: A Laboratory Manual (We all have this on our bookshelf, yes?) The idea that there were reliable "standard procedures" that could ensure success was farcical—an understatement that other scientists understand as code for "we spent years getting this to work; good luck figuring it out ;)."
So, as an appreciation of what it takes to make progress on the frontiers of science, here is approximately what’s involved.
Prerequisites: Dexterity, Glassblowing, and Zen Mastery
Do you have what it takes to master transgenic mouse creation? Well, do you have the dexterity of a neurosurgeon? Because you’ll be micro-manipulating fragile embryos with the care of someone defusing a bomb—except the bomb is smaller than a grain of sand, and you need to keep it alive. Have you trained in glass-blowing? Hope so, because you’ll need to handcraft your own micropipettes so you can balance an embryo on the pipette tip. Yes, really.
And most importantly, do you sincerely believe that outcomes are irrelevant, and only the endless, repetitive journey matters? If so, congratulations! You may already be a Zen master, which will come in handy when you’re objectively failing your boss’s expectations every single day for what feels like an eternity. Success, when it finally comes, will be indistinguishable from sheer, dumb luck, but the stochastic randomness won’t stop you from searching frantically through your copious notes to see if you can pinpoint the variable that made it finally work!
Let’s go a little deeper so we can understand why the Games team's achievement was considered so monumental—and why almost everyone viewed the results in the best possible light.
The Science and Alchemy of Designing the Perfect Genetic Construct
First, these researchers needed to design a genetic construct. What's a construct, you ask? It's a carefully engineered piece of DNA that harnesses circular plasmids (tiny rings of DNA naturally found in bacteria) to introduce foreign genes into mammalian cells. Through a painstaking process called sub-cloning, equal parts molecular biology and divination, they managed to insert into their mouse a human APP gene carrying the mutation found in families with high rates of early-onset Alzheimer's.
You can design your construct perfectly on paper, but in truth, you solve the problem by tweaking reagents like an alchemist, trying to find the perfect brew to coax your foreign gene into the plasmid at high efficiency.
To be considered a valid Alzheimer’s model, the Games mouse needed to express human APP at levels high enough to cause Alzheimer's-like pathology. Previous attempts by other labs had yielded mice that showed little to no amyloid plaques. Scientists suspected that higher expression levels might overcome this hurdle. They introduced the PDGF-β promoter, a genetic “on switch” that controls when and where a gene is activated to drive high expression in neurons; they included introns in the construct to allow for alternative splicing, a process that enables cells to produce different versions of a protein, in this case ensuring expression of the full range of amyloid-beta peptides seen in human Alzheimer’s.
But even with these clever designs, they had almost no control over where their transgene would integrate into the genome, how many copies would insert, or how much gene expression they’d elicit.
Microinjection to Model Organism: The Birth of Transgenic Line #100... #101... #102... Are We There Yet?
When the Games team finally (miraculously!) had a perfect construct, the next phase began: obtaining precisely timed mouse embryos. To make this transgenic mouse line, researchers needed to inject the transgene into single-cell fertilized embryos, prior to the first cell division event. It’s a very small needle, but only by threading it can you ensure that the transgene incorporates into the DNA of every dividing cell. Back when the study was conducted in the 1990s, the Games team had to rely on natural fertilization, meaning they needed female mice that had just ovulated and mated.
Thinking about this work triggers me. I spent years of my PhD setting up and monitoring mouse breeding pairs for timed pregnancies. Every morning began with the ritual of checking for copulatory plugs (don’t ask!). Only ~20-25% of pairs would successfully mate overnight: some females aren’t receptive; some males are layabouts. The failed pairings had to be separated and re-paired in the evening, so fertilization timing could be precisely tracked. Once mating was confirmed (those copulatory plugs again), the female was euthanized, and her oviducts—tiny tubes containing the precious fertilized eggs—carefully dissected. Then you flush out the one-cell zygotes using a finely-tuned glass pipette (yet another moment where glass-blowing skills came in handy).
Now for the hard part: microinjection—the insertion of the transgene into an egg the diameter of a human hair (you really should watch the video). You need the steadiness of a bomb defuser, the aim of a sniper, and just enough pressure to get the DNA inside the pronucleus without rupturing the egg (because ruptured eggs = weeks of work wasted instantly). All this while the eggs had to be kept alive under precisely controlled conditions. Today, fancy $100K integrated microinjection/scope systems help the process along (though it still takes years to master), in the early 1990s, microinjection was brutal—for all parties. Only a handful of scientists in the world could perform it with any consistency.
Let’s pause to acknowledge the obsessive craft of the skilled bench scientist.
The PDAPP Mouse Arrives
Once injected, the embryos were surgically transferred into surrogate mothers, then scientists waited anxiously for 18–21 days to see if any pups survived. When they did, DNA was extracted, and tests were run to see which, if any, carried the transgene. Success rates? Single-digit percentages. For every founder animal that carried the transgene, there were at least an order of magnitude more failed attempts.
The effort layered chance upon pure chance—literally hoping that the DNA randomly integrated into the genome. Where? Unknown. How many copies? Uhh. Would it express properly? Flip a coin.
That’s what made the PDAPP (Platelet-Derived growth factor (PDGF-β) Amyloid Precursor Protein) mouse a remarkable achievement. When it finally worked and was replicable in the creators’ lab, it wasn’t just a technical success—it was a miracle of molecular biology and tenacity.
How to Read a Scientific Paper
Actively.
Imagine you’re a molecular neuroscience grad student in 1995. You’ve just sat down at your bench in the morning when your PhD supervisor calls you into her office. She’s at her desk, her hand pressed down on the latest issue of Nature like she’s trying to keep it from flying away.
“They did it. They made a transgenic Alzheimer’s mouse that shows the pathology. Go print color copies for everyone in the lab, then bring this back to me. You’ll present it in lab meeting tomorrow.”
You have 24 hours to get ready to lead journal club on the biggest translational neuroscience story in years. What do you do?
Crushed for time, most scientists I know read papers passively. They start at the beginning and work their way to the end, following the path the authors laid out for them. They become susceptible to the trap Medawar and Bernard warned about: mistaking the arc of a narrative for genuine logical coherence.
To see the substance through the argument in the Games paper, you’ll need a more active, detective-like approach. If you’re going to be convinced, you need to decide before you read what it will take to convince you. So, you begin with what may be the most important task of active reading: before looking at the paper, you imagine the experiments and results that would justify the claim that amyloid causes Alzheimer’s—and that the matter is settled.
What would we need to see to be convinced? Let's apply some key principles of experimental design:
Temporality: If amyloid beta (Aβ) drives pathology, plaques should appear first, followed by neurodegeneration, then cognitive deficits.
Sufficiency [2]: If Alzheimer’s-like pathology can result from ramping up human APP expression alone, then the PDAPP mouse is a quasi-test of sufficiency. If this mouse model develops plaques, tangles, synaptic loss, neurodegeneration, and cognitive impairment, then Aβ might be sufficient to initiate the disease process.
Necessity: An even stronger case for the amyloid cascade hypothesis requires showing that Alzheimer’s pathology can’t develop or progress if Aβ is absent or blocked.
Mechanism of Action: A truly convincing paper should demonstrate the biology of how Aβ triggers neurodegeneration.
Critical Controls: Including controls in your experiments helps to demonstrate that your predicted effect isn’t arising for some other reason. Here are some controls you’d like to see in the PDAPP mouse study if you’re going to be convinced of the amyloid cascade hypothesis:
Absence of plaques in mice not engineered to overexpress APP.
Comparisons between brain regions with and without plaques. If you see neuronal death, it should co-occur with the presence of plaques, not be spread throughout the brain.
Evidence that APP overexpression isn't driving toxicity, which can cause similar damage to the brain. This is very hard to control for, so the best course of action is to try to match human APP expression levels in your mouse model.
Controls are an indispensable reality check for strong inference—the practice of designing experiments that not only test a hypothesis but aim to disprove it or eliminate alternatives. The concept was introduced in 1964 by the biophysicist John Platt, who observed that some scientific fields advance rapidly while others stagnate. The difference, he argued, wasn’t in the complexity of the problems or the brilliance of the researchers, but in the systematic use of what he called strong inference.
Unlike the traditional scientific method, which often tests a single hypothesis against a null, strong inference begins with multiple competing explanations. It then designs experiments specifically intended to rule them out. Over time, this produces a branching tree of narrowing possibilities, steadily eliminating what doesn’t hold up.
This approach also guides how we read. Asking what control would disprove the claim—or what alternative wasn’t tested—is the core of strong inference.
Detective Work—Decoding the Paper
With our backstory in hand and analytical toolkit in mind, let’s see if the PDAPP mouse delivers on the amyloid cascade hypothesis.
It’s time to interrogate the key figures. In a paper like this, the figures are the empirical backbone of the argument. Of course, the authors have carefully chosen what to show and how to frame it. Our job is to assess whether the evidence supports their claims.
Figure 1: Confirming the PDAPP Mouse Expresses Human APP
Big picture message: The authors successfully engineered a mouse with a functioning human APP transgene. While Figure 1 has 5 parts, Panel d (below) is where we get the clearest confirmation that the human mutant APP transgene integrated itself into the mouse genome and produced Aβ in the expected location in the mouse brain. Stare at Panel d for a few seconds and then we’ll talk about it.
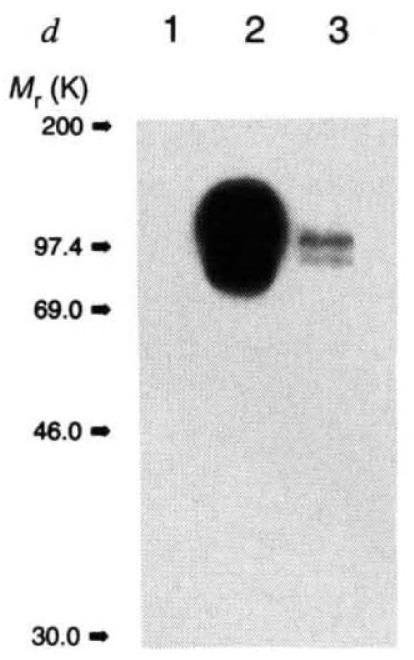
What the Heck is Going on in Panel d?
Panel d is an immunoblot (Western blot), a technique that tells us whether a specific protein is being produced in a sample. The figure compares human amyloid precursor protein expression in samples from three brains, corresponding to the three “lanes” shown along the top: a normal mouse (Lane 1), a mouse with the human APP transgene (Lane 2), and a human who had Alzheimer’s disease (Lane 3). The blots (the bands and blobs) are the data.
For purposes of this figure, proteins can differ in two important ways:
They can differ in molecular weight (size), scored on the scale along the vertical axis on the left.
They can be expressed to a greater or lesser degree, reflected in the intensity (darkness and spread) of the bands. Lighter means less protein expression, darker more. (Note: It’s fine that there are two bands in lane 3).
Let’s pause to consider what we should expect to see in this blot if the PDAPP mouse is to be considered a reasonable model of human Alzheimer’s disease. Two things!
Ideally, lane 1 (normal mouse) will be empty; and
The PDAPP mouse sample in lane 2 will have the same molecular weight (height on vertical axis) and expression level (intensity and spread) as the human sample in lane 3.
Why do we want to see these results? It matters because our ultimate goal is to develop treatments that work in people, not just in mice. To make that leap from a mouse model, the PDAPP mouse needs to replicate the key features of Alzheimer’s disease in humans—not just produce APP but drive similar amyloid-induced disease processes in the brain.
Ok, so on point one, ✔ — nothing in lane 1.
Point two? ❌ While the human sample has distinct bands in lane 3, the PDAPP mouse in lane 2 appears as a giant, smeared blob. Are these the same protein size? Impossible to tell!
This happens because, relative to the human sample, the PDAPP mouse is drowning in APP—at least 10 times more, according to the paper’s text, and possibly much more by eye. When you do a Western blot, you set an exposure time for your image, just like with a manual camera: too short an exposure, and faint bands won’t appear; too long, and strong signals become an oversaturated mess. Here, no single exposure could produce similar-looking PDAPP and human samples. It’s like trying to take a photo of a candle next to the sun—you can’t adjust for both at once.
A proper Western blot should show clean bands to confirm protein size and check for unexpected degradation products, but this overloaded mess makes it impossible to tell whether APP is being processed normally. A Western blot like the one shown in Panel d usually indicates either sloppy technique (overloading the gel, overexposing) or a fundamental issue with the model itself (massive expression differences between samples).
The fine print explains why: the PDAPP mouse carries 40 copies of the APP transgene, all inserted at a single site in the genome. For context:
✔ At most, humans have 2 copies of APP (one from each parent).
✔ PDAPP mice have 40 human copies—plus their 2 normal mouse copies.
I’m sure this blot led to high-fives in the lab—earlier models struggled to express APP at all, so getting massive overexpression must have felt like a breakthrough.
But now I’m worried.
If we’re trying to create a human-comparable Alzheimer’s model, this much APP might be way too much. Why might this be a problem?
APP expression at this level doesn’t mirror expression levels in human Alzheimer’s. Alzheimer’s patients don’t have 40 copies of APP. If it takes this much overexpression (and a mutant form at that!) to drive pathology in mice, are these mice even an appropriate animal model for Alzheimer’s?
Excess protein can stress neurons in ways unrelated to amyloid. What if the brain is freaking out not because of Aβ toxicity, but because it’s drowning in APP?
Overexpression alone isn’t a dealbreaker. Many successful transgenic models for cancer, Huntington’s, and Parkinson’s disease rely on high gene expression to accelerate pathology and make the disease more tractable for study. These models have been invaluable for understanding disease mechanisms and testing therapies. So, while the extreme APP overexpression in PDAPP mice raises concerns, it doesn’t automatically invalidate the model—we could still be on the right track.
But your scientific spidey sense should be tingling. If the pathology we’re about to see is simply a side effect of astronomical overexpression, then this mouse may be modeling an extreme artifact, not human disease—and there aren’t easy ways to tell the difference.
Let’s keep going…
Figure 2: We have amyloid plaques! (I think?)
Figure 2 is supposed to convince us that the amyloid plaque burden in PDAPP mice matches that of the human brain and that it worsens over time, just like in Alzheimer’s disease. But instead of giving us clear, apples-to-apples comparisons, the figure presents a frustrating mismatch of images that makes it difficult to draw meaningful conclusions.
First, let’s talk about the comparison of PDAPP plaques to human Alzheimer’s brain plaques. The authors claim that the amount of amyloid in 9-month-old PDAPP mice is equivalent to what’s seen in humans, but the figures they provide make this impossible to assess. Figure 2a shows a large image of a PDAPP mouse brain with a tiny inset of a human Alzheimer’s brain, which, at a glance, actually looks like it has way more plaques. But because of the size difference of the images (not the magnification; magnification is the same; see the horizontal bars in each image) and lack of anatomical markers on the inset, we can’t visually compare them.
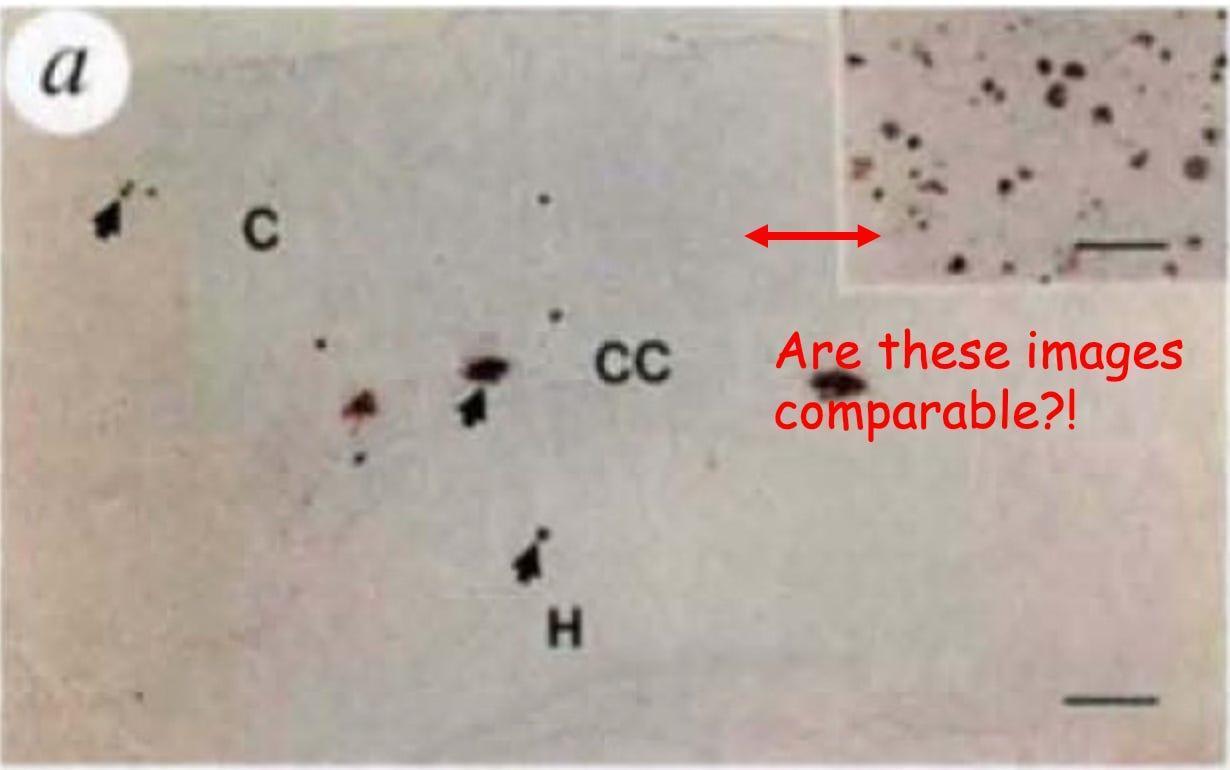
The obvious fix? Show an image of the human sample that is the same magnification and size as the mouse so we can actually evaluate the claim. I can’t think of a justification to do it any other way. Give us a direct, side-by-side comparison to human pathology. Full stop.
While I won’t reprint the Figure 2 subfigure here, to support a claim that PDAPP mouse plaque load increases over time as in human Alzheimer’s, the authors use images with inconsistent size as well as magnification! Do mouse plaques increase over time? We can’t be sure.
In summary, Figure 2 systematically makes it really hard to:
Compare plaque pathology in PDAPP mice to human Alzheimer’s brains.
Evaluate whether mouse amyloid progresses over time in the Alzheimer’s way.
It’s frustrating because these are the key questions Figure 2 is supposed to answer. Grrr.
Figure 3: What’s in These Plaques?
Figure 1 confirmed that the PDAPP mouse makes human APP and a lot of it. Figure 2 tried (somewhat unsuccessfully) to convince us that PDAPP mouse plaque burden mirrors human Alzheimer’s. Figure 3 shifts focus to the composition of those plaques. This is important because not all amyloid deposits are equal—some forms of Aβ are more toxic, more structured, or more likely to trigger downstream pathology than others.
So, what do we learn? The PDAPP plaques aren’t just random protein aggregates; they contain key molecular features of Alzheimer’s pathology.
Panels d & e: Do These Plaques Act Like Human Plaques?
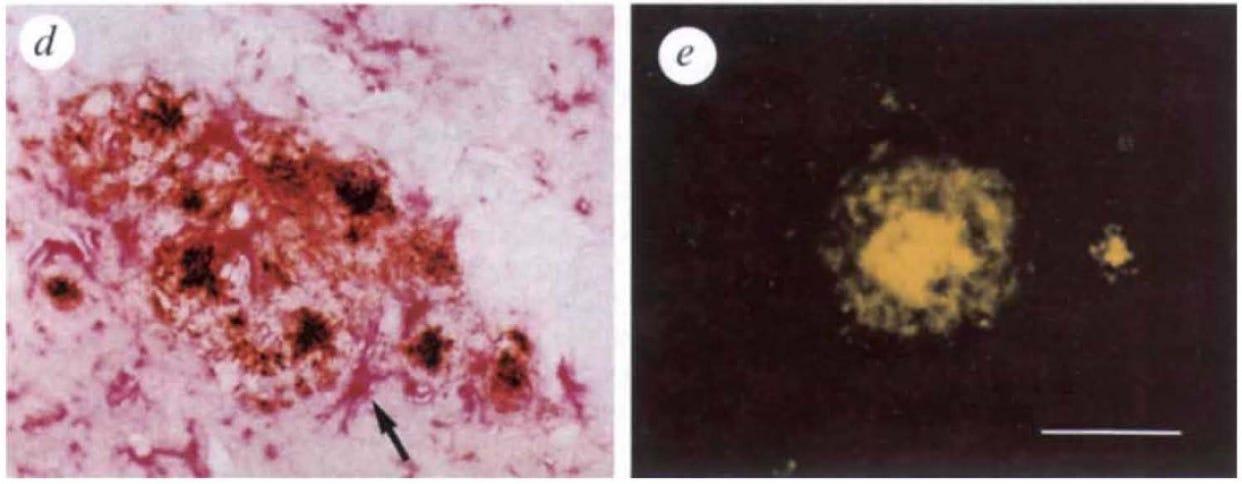
✔ Panel d shows PDAPP mouse astrocytic gliosis (see arrow pointing to the angry looking red thing), a type of inflammation where one kind of brain cell (astrocyte) clusters around plaques. Gliosis is a hallmark of neuronal damage in human Alzheimer’s, suggesting that the plaques in PDAPP mice and humans are similarly biologically active.
✔ Panel e reveals that PDAPP mouse amyloid deposits are similar to human Alzheimer’s plaques in two other ways.
Both have plaques that form structured, compacted fiber strands (fibrils), causing them to stain positive for Thioflavin-S.
The larger of the two mouse plaque deposits in Panel e shows a dense core with a surrounding halo, a hallmark of amyloid plaque deposits in human Alzheimer’s.
So far, so good—amyloid plaques in PDAPP mice aren’t just amorphous protein junk; they’re structured, biologically reactive, and surrounded by gliosis.
But Wait… Something’s Missing.
The other half of Alzheimer’s signature pathology is neurofibrillary tangles (NFTs). Plaques and tangles are the Batman and Robin of Alzheimer’s disease: where plaques go, tangles are supposed to follow.
So, where are they?
Search all you want. You won’t find a single one. Instead, buried in the text, we get this:
“Preliminary attempts to identify neurofibrillary tangles … were negative, consistent with their well-known absence in rodent tissues.”
This is a devilish bit of phrasing. The conditional language here—“consistent with their well-known absence”—makes it sound like no one expected to see tangles in the first place. Really? The commentary accompanying the paper also hedges:
“It is likely that those who are skeptical of the amyloid-cascade hypothesis will draw comfort from the apparent absence of tangles in the mice… However, it remains possible that their absence reflects the fact that they are merely a marker of cell injury …: indeed, in some cases of dementia, neuritic plaques occur without tangles, meaning that tangles may indeed be epiphenomena.”
My Translation: The amyloid cascade camp was absolutely hoping to see tangles! Their absence wasn’t expected—it was disappointing. But instead of confronting what this means for the mouse model, the paper and commentary don’t just shift the goalposts, they suggest taking them off the field. They’re using an unvalidated mouse model—fresh off the bench—to call for sweeping change in our fundamental understanding of the biology of Alzheimer’s disease in humans.
Come now! This is completely backwards. When a mouse model fails to produce a core feature of the disease, it’s time to own the potential limitations, not reinterpret the disease to fit the model. 🤯
So where does that leave us? For now, your skepticism of the PDAPP Alzheimer’s model and the amyloid cascade hypothesis itself should be growing. The PDAPP mouse makes amyloid, those plaques look structurally realistic, and they trigger gliosis-type inflammation. But there’s no tau pathology—no tangles—despite their strong link to neuronal dysfunction and cognitive decline in human Alzheimer’s. In retrospect, we now know that the absence of tangles in these mice foreshadowed a key limitation of amyloid-based models.
On to Figure 4. . .
Figure 4: The Gap Between Plaques and Neurodegeneration
The final figure is meant to show how amyloid plaques impact neurons in the PDAPP mouse, with comparisons to human Alzheimer’s disease.
I’m only going to show Panel h from Figure 4, but let’s briefly talk about the other panels. The authors examine neuronal structure, highlighting neuritic damage, synaptic loss, and cellular stress. Using images from a confocal microscope, they demonstrate distorted neurites (aka stressed-out neurons) surrounding plaques in both human Alzheimer’s and transgenic mice and reduced synaptic density and dendritic markers in the PDAPP mouse compared to mouse controls. All good stuff.
Finally, Panel h reveals abnormal neuronal structures near amyloid deposits. Let’s take a closer look at the highest resolution image, captured with an electron microscope. Electron microscopy can reveal the fine details inside cells, down to individual organelles. If plaques are truly destroying neurons, this is where we should see the damage up close.
Here it is.
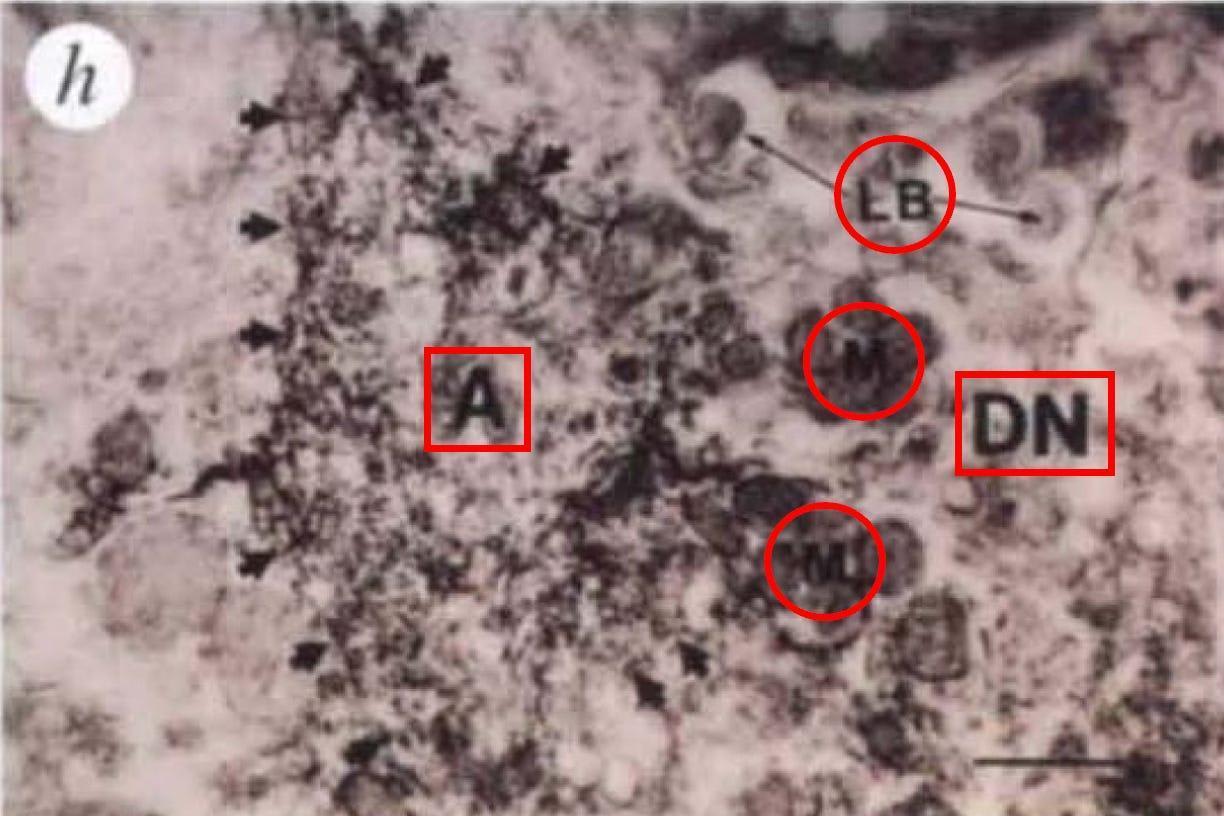
At first glance, it seems to provide exactly what we'd expect in an Alzheimer's model: an amyloid deposit (A) sitting next to a dystrophic neurite (DN), which contains swollen, abnormal mitochondria (M) and dense bodies (LB), all signs of cellular stress. These kinds of metabolic defects—like disrupted mitochondria that can no longer generate cellular energy and accumulated protein aggregates that clog cellular machinery—have been observed in human Alzheimer’s brains where neurons are in distress.
But there's a catch. This is one neurite. At this image resolution, if plaques were sufficient to drive large-scale neurodegeneration, we'd expect to see widespread cellular destruction, not just a single distressed process. In human Alzheimer’s brains, electron microscopy images often show fields of degenerating neurons, ruptured organelles, and catastrophic synaptic breakdown. Yet here, we get a highly selective image of one damaged neurite, without any indication of how representative this is across the brain.
This raises an important question: Is amyloid actually killing neurons in this model, or are neurons adapting to the presence of plaques? If widespread cell death were happening, this is the figure where it should be most obvious—but instead, we see only localized damage.
This discrepancy matters because in human Alzheimer's disease, cognitive decline correlates most strongly with neuronal loss (cell death), not with plaque burden. Some patients with significant amyloid deposits show minimal symptoms; others, with fewer plaques but more neurodegeneration, experience severe dementia.
Panel h is meant to reinforce the case that Aβ drives neurodegeneration, but it may instead highlight a key limitation of the PDAPP mouse: neurons near plaques look stressed, but they aren't dying in droves. Why aren’t we presented with evidence of more devastation at this scale?
Revisiting Our Expectations
Did the PDAPP Mouse Deliver on Its Bold Claims?
We’ve spent this deep dive critically analyzing the key figures and missing evidence. Now it’s time to step back and ask: Did this paper adequately support its sweeping conclusions?
The authors make a confident claim in their discussion:
“Our transgenic model provides strong new evidence for the primacy of APP expression and Aβ deposition in [Alzheimer’s] neuropathology and offers a means to test whether compounds that lower Aβ production and/or reduce its neurotoxicity in vitro can produce beneficial effects in an animal model prior to advancing such drugs into human trials.”
Likewise, the accompanying commentary framed this paper as a game-changer, declaring that the long-standing debate over amyloid’s role in Alzheimer’s was now effectively settled.
But were we convinced?
Fortunately, at the start of this analysis, we took the time to define the experimental standards needed to evaluate these claims. We don’t have to rely on gut feeling or rhetorical framing to decide. Our approach—laying out our expectations in advance—gave us the tools to spot what the paper shows and what’s absent but essential.
Flip the Script
The most powerful scientific minds possess a talent for inverting problems. Instead of asking whether this paper supports the amyloid hypothesis, we can ask whether the hypothesis was undermined? This intellectual jiu-jitsu is the essence of Platt's strong inference method.
Imagine that the Games team’s hypothesis and experimental results landed on your desk in raw form, without the narrative of the paper or the triumphant accompanying commentary. Set aside for a moment your appreciation for the remarkable transgenic technical feat—precisely inserting a human gene into a mouse's genome and having it produce a functional protein and a bunch of amyloid plaques. Might you reach different conclusions?
If amyloid truly drove neurodegeneration, these mice—riddled with plaques—should have shown devastating neuronal death. Instead, the neurons looked stressed but largely intact, their organelles preserved. Plaques without consequence. Smoke without fire.
If the cascade hypothesis was correct, plaques should trigger tau pathology, producing the neurofibrillary tangles seen in human Alzheimer’s. Yet despite astronomical amyloid levels, the PDAPP mice developed no tangles at all. The chain of causation broke mid-link.
If this was truly a disease model, it should mirror natural conditions. Yet creating these mice required inserting forty copies of a mutant APP gene, producing protein levels at least ten-fold higher than any human Alzheimer’s patient. This wasn’t a model of disease. It was a model of artificial protein overload.
Moreover, and perhaps most tellingly, the paper included no behavioural or cognitive testing. Alzheimer’s devastates patients not because of plaques, but because of profound memory loss and cognitive decline. Did these mice develop memory problems? Did their cognitive function deteriorate over time?
In a model of Alzheimer’s disease, behaviour is a critical test. It shows whether the brain changes we’re studying lead to symptoms like memory loss or confusion. Of course, mice don’t behave like humans, but they can show species-appropriate changes—like trouble navigating a maze—that reflect similar brain disruptions.
Those in positions of power and authority either failed to see these flaws or chose to overlook them. What happened next was astonishing in its speed and scale.
Within a year, Athena Neurosciences, where Games and his colleagues worked, was acquired by Elan Corp. for $638 million. In the press release, Elan declared the acquisition “an opportunity to capitalize on an important therapeutic niche,” combining Athena’s “leading Alzheimer’s program” with Elan’s drug development pipeline. The PDAPP mouse had transformed from laboratory marvel to the cornerstone of a billion-dollar strategy. The industry followed. Pharmaceutical giants, biotech startups, research foundations—all placed their bets on amyloid.
One by one, those bets failed. By the time Elan collapsed in 2013, it had sponsored four failed Alzheimer’s drugs, hemorrhaging more than $2 billion in the process. Some trials caused patients significant harm.
The Games paper, read carefully and critically, hinted at its flaws. They were there waiting for anyone who cared to look beyond the polished narrative.
Those concerns we raised about the model? The wild APP overexpression, the absence of tangles, the neurodegeneration—they weren’t just theoretical issues. Over the following years, a series of studies confirmed that these flaws fundamentally disqualified the PDAPP mouse as a reliable model of Alzheimer’s disease. And when behaviour was finally tested, the results raised more concerns than confidence. Memory problems showed up before plaques had formed, and didn’t progress in the way human symptoms do. Instead of validating the model, the behaviour suggested that the brain had been disrupted by the artificial overexpression itself—not by the pathology the model was meant to study.
And yet, the story held for decades. In many places, the amyloid cascade hypothesis remains entrenched to this day. Its staunchest defenders still occupy some of the most influential positions in research institutes, scientific societies, and grant review panels. Under their influence, evidentiary standards were shifted. Assumptions, and even the diagnostic criteria (!), were revised to accommodate half-satisfactory results, rather than to face falsification. Correlations were elevated to causes. And over time, the elegant machinery of scientific inference began to slip its gears. The field can sometimes feel like it’s circling endlessly round a well-funded cul-de-sac—exhausting resources while alternative ideas remain unfunded, unpursued, or unheard.
The amyloid cascade was a great hypothesis, worthy of testing, and more importantly, of scrutiny. Its most intransigent defenders might do well to recall another bit of Sir Peter Medawar’s wry clarity:
“The intensity of the conviction that a hypothesis is true has no bearing on whether it is true or not.”
At some point, science’s self-correcting machinery—and the brilliance and curiosity of a new generation of researchers—will win out. It is time to widen the lens.
Endnotes:
[1] I keep name-dropping journals (almost always Nature and Science) and realize that this means little to most people. Nevertheless, having a passing understanding of the tiers of academic publishing is part of scientific literacy.
If you've ever wondered why scientists scramble to publish in Nature, Science, or Cell, think of them as the holy trinity of scientific prestige, each with its own personality. Nature and Science were established in the late 1800s—Nature published by the Brits and Science by the American upstarts. Cell is the newcomer; established in 1974 in Cambridge, Massachusetts.
In terms of temperament, Nature is the flashy cosmopolitan—broad, attention-grabbing, and often favouring "sexy science" that makes headlines (black holes, CRISPR, or ancient human fossils). Science is the serious, translationally-minded intellectual—rigorous, respected, and slightly less obsessed with media hype (CRISPR before it was famous). Then there's Cell, the molecular biology workhorse, where groundbreaking discoveries in pre-clinical work are dissected in exquisite mechanistic detail (if you love signaling pathways, this is your jam). Publication in any of these journals is the scientific equivalent of winning an Olympic medal (or at least making an Olympic team, depending on your position in the author list).
One tier down, you'll find specialty journals like Neuron, Nature Neuroscience, and The Journal of Clinical Investigation (and reams of others for specific fields), which publish longer, more methodically comprehensive studies. I tend to prefer reading papers from these journals as they provide greater detail and present more fully developed work. These papers may not be as "hot off the press" or media-friendly, but they often demonstrate greater scientific rigor and better withstand the test of time.
Meanwhile, in medicine, The Lancet and The New England Journal of Medicine (NEJM) tower over most others with impact factors of 98.4 and 96.2, respectively (generally, the higher the impact factor the greater the prestige). This reflects their enormous readership—there are far more medical doctors than PhDs. But at the top, it’s CA: A Cancer Journal for Clinicians, with a staggering 286.13 impact factor, a reminder of cancer’s toll and where our research priorities and funding are concentrated.
[2] A brief note on terminology: I use the terms “necessity” and “sufficiency” here as they were traditionally understood by molecular biologists in the 1990s.
Strictly speaking, the terms originate in formal logic and philosophy, where they have precise meanings related to logical entailment: if P is sufficient for Q, then whenever P is true, Q must also be true; if Q is necessary for P, then P cannot be true unless Q is also true. These relationships are logical, not causal.
In experimental biology, however, the terms have been adapted into a more practical shorthand. A molecule is often described as “necessary” if removing it disrupts a biological process, and “sufficient” if adding it can induce or mimic that process. But this looser, causal usage rarely matches the strict logical rigor the terms imply—and it can obscure complexity when used uncritically in systems with many interacting factors. Some have proposed retiring or replacing these terms altogether in favour of more nuance.
Nevertheless, for the purposes of this review (and to remain faithful to how the field understood these concepts at the time) I use “necessity” and “sufficiency” in the conventional experimental sense: as shorthand for causal roles a factor was believed to play—whether it could trigger a process or was required to sustain it.
