
DRUGAI
今天为大家介绍的是来自美国斯克里普斯研究所的Douglas J. Kojetin团队的一篇论文。核受体(Nuclear receptors, NRs)是一类能在转录激活和抑制两种状态之间切换的蛋白质,这种状态转换可以通过与特定分子结合来稳定。目前大多数核受体调节分子要么能激活转录,要么保持中性,很少有能够抑制转录的反向激动剂,这使得我们难以全面理解核受体的作用机制。本研究发现了一系列能调节过氧化物酶体增殖物激活受体γ型(PPARγ)的分子,这些分子的作用范围从抑制到激活都有覆盖,通过对其结构进行细微调整就能改变其功能。研究团队通过晶体结构分析观察到了完全抑制状态下的PPARγ,并利用核磁共振波谱和构象-活性关系分析发现,这些调节分子能够引导PPARγ在不同功能状态之间转换,而这些状态在自然状态下本就存在。这一发现为我们提供了一个重要的分子设计框架,帮助我们理解如何通过最小的化学结构改变来增强PPARγ的抑制效果,并揭示了这些改变是如何影响PPARγ的动态变化的。

这项研究深入探讨了核受体(Nuclear receptors, NRs)这类重要的蛋白质,它们约占FDA批准药物靶点的15%。研究特别关注了过氧化物酶体增殖物激活受体γ型(PPARγ),这是一种能感应脂质的核受体,也是抗糖尿病药物的作用靶点。研究发现,PPARγ的药物结合区域在自然状态下会在活性和抑制两种状态之间自由转换。科研团队重点研究了两种化合物:T0070907和GW9662。这两种化合物都具有相同的基本结构—2-氯-5-硝基苯甲酰胺(2-chloro-5-nitrobenzamide),但通过调整其中的酰胺基团,可以改变它们对PPARγ的调节作用。通过晶体结构分析和核磁共振技术,研究人员发现对T0070907进行细微的化学修饰,可以得到一系列活性不同的化合物,从完全抑制到完全激活都有。这些化合物通过影响PPARγ的空间结构来调节其功能,为开发新型药物提供了重要的理论基础。特别值得注意的是,这类化合物正在被开发用于治疗PPARγ过度活化的膀胱癌。
提出假设并验证以增强抑制效果
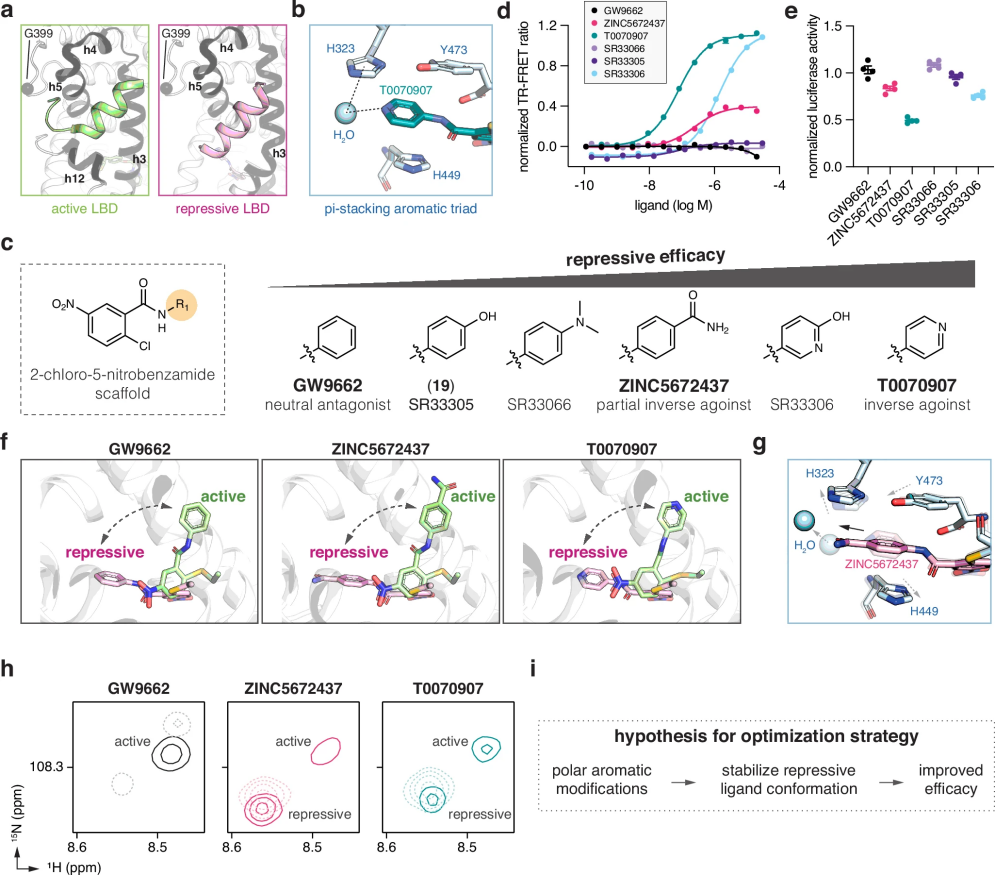
图 1
如图1所示,研究团队揭示了PPARγ蛋白质如何被不同的化合物调控。在正常激活状态下,PPARγ的关键结构(螺旋12)位于蛋白质表面(图1a)。而当抑制剂T0070907结合时,它能与蛋白质中的三个特定氨基酸形成稳定的相互作用,同时通过一个水分子形成额外的连接(图1b)。基于这一发现,研究人员提出假设:含有特定极性结构的化合物可能通过类似的作用方式来增强抑制效果(图1c)。他们通过两种实验方法(图1d和1e)验证了这一假设,并发现一个名为ZINC5672437的化合物具有较好的抑制活性。
晶体结构分析显示,研究的三种化合物都能在蛋白质中采取两种不同的结合方式(图1f)。其中,ZINC5672437通过其特殊的化学结构改变了原有的水分子连接方式(图1g)。核磁共振实验进一步证实,ZINC5672437能够诱导PPARγ形成两种稳定的构象(图1h)。最终,研究结果表明,在化合物结构中引入特定的极性芳香基团,可以有效地稳定PPARγ的抑制构象,从而提高其抑制效果(图1i)。这一发现为开发新型PPARγ调节剂提供了重要的设计思路。
设计一系列配体以探究激活范围与抑制效果
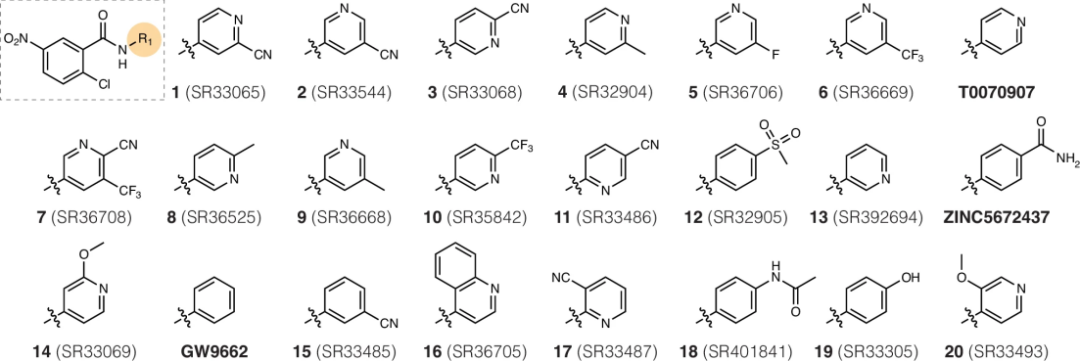
图 2
研究团队设计了一系列化学结构相似但功能各异的化合物,用来研究它们对PPARγ蛋白质的调控作用。这些化合物都基于2-氯-5-硝基苯甲酰胺结构,通过在特定位置添加不同的化学基团来改变其功能(图2)。
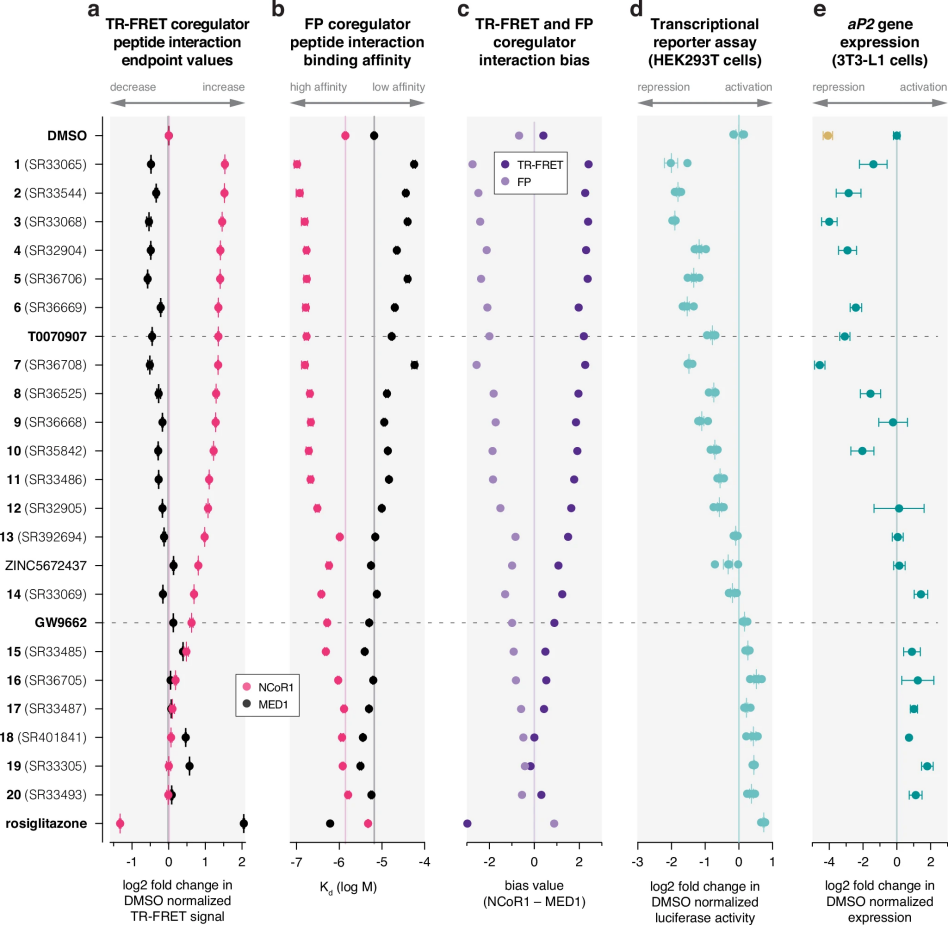
图 3
图3呈现了多维度的实验数据分析:
(a) 通过时间分辨荧光共振能量转移(TR-FRET)实验,研究这些化合物如何影响PPARγ配体结合域(LBD)与辅抑制因子NCoR1和辅激活因子MED1的相互作用。
(b) 采用荧光偏振(FP)技术直接测量结合亲和力。
(c) 计算了协调因子偏向性,反映NCoR1和MED1在TR-FRET效能和FP亲和力方面的差异。
(d) 通过荧光素酶报告基因实验评估化合物对PPARγ转录活性的影响。
(e) 在3T3-L1前脂肪细胞中检测了PPARγ靶基因aP2/FABP4的表达变化。
实验结果显示,这些化合物的活性范围很广,从完全抑制到部分激活都有。含有特定极性结构的化合物表现出更强的抑制作用,而那些含有疏水性结构的化合物则倾向于表现出激活作用。
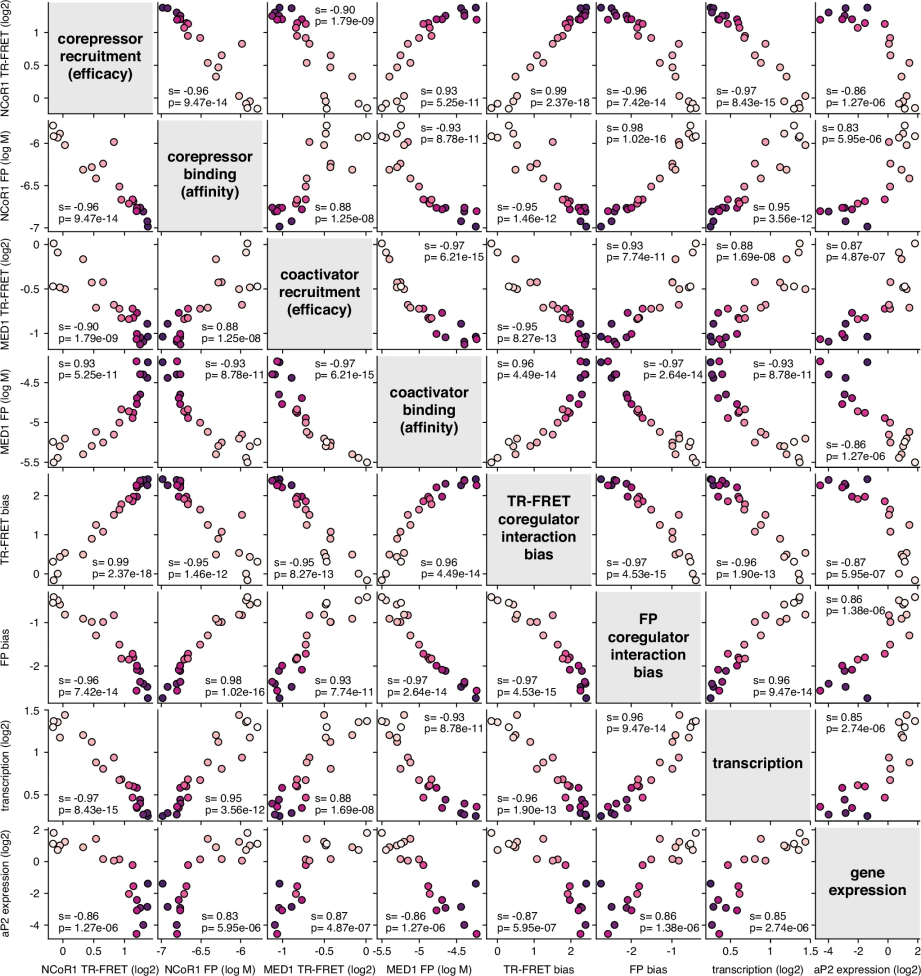
图 4
图4通过斯皮尔曼相关系数分析显示,生化实验数据与细胞功能表现高度相关。转录抑制和aP2表达降低与NCoR1结合增强呈正相关,与MED1结合减弱呈负相关。这表明使用纯化PPARγ LBD蛋白的384孔板生化实验可以有效预测这类化合物对完整PPARγ转录活性和脂肪生成过程中靶基因表达的影响。
晶体结构揭示改进型辅抑制因子选择性效能的分子机制
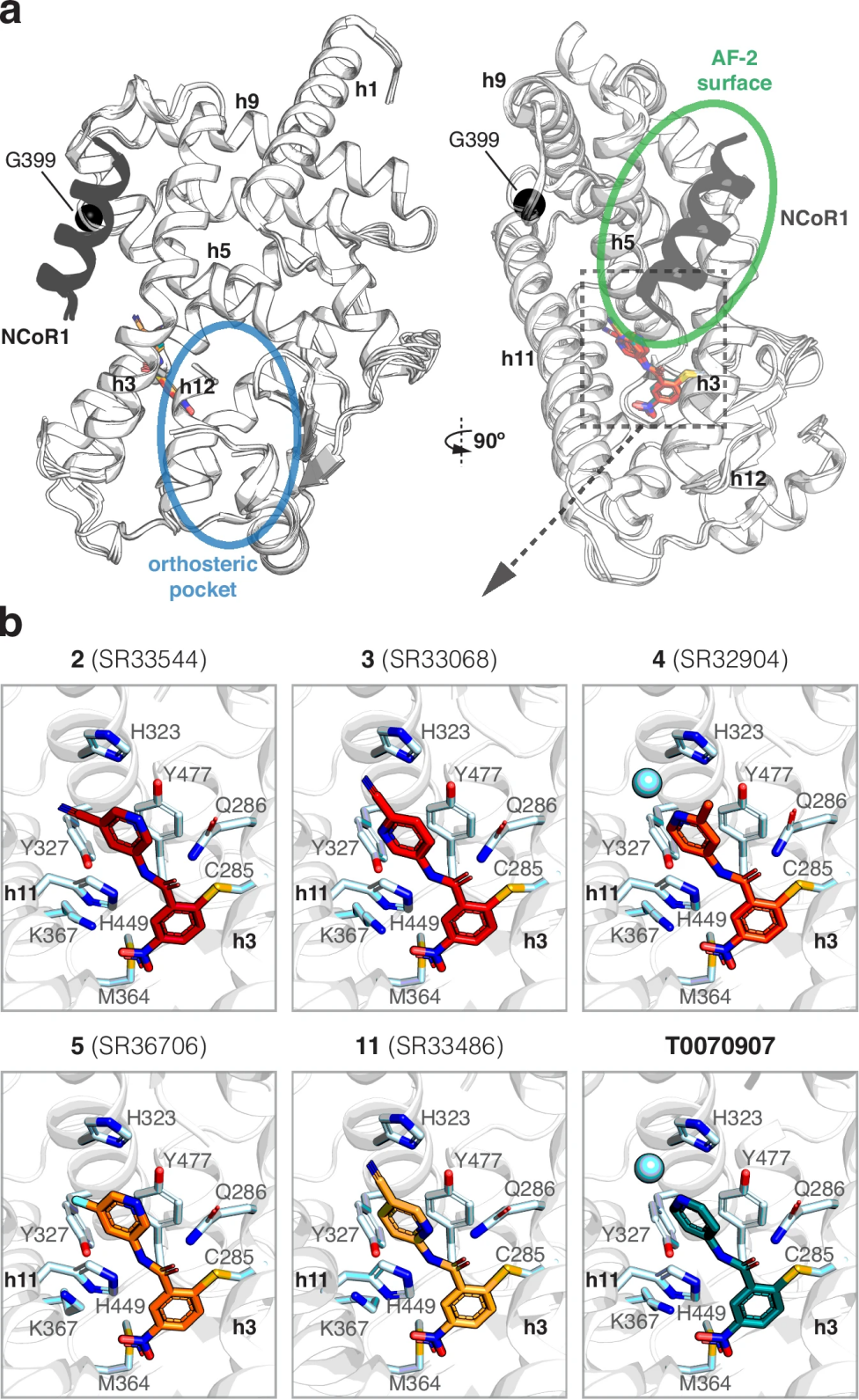
图 5
图5通过晶体结构揭示了改进型反向激动剂的分子作用机制。图5a展示了PPARγ配体结合域与NCoR1辅抑制因子肽及多个化合物的复合物晶体结构。这些结构具有以下特点:
图5b详细展示了化合物与受体结合位点的相互作用模式:
量子力学计算进一步证实,这些改进型化合物与受体的结合能比原型化合物T0070907更有利,这可能源于它们优化后的极性相互作用。不过,结合能的强弱并不完全对应于化合物的活性,说明还存在其他影响因素,如水分子介导的相互作用或化合物的构象刚性等。
配体改变了功能性的LBD构象系综
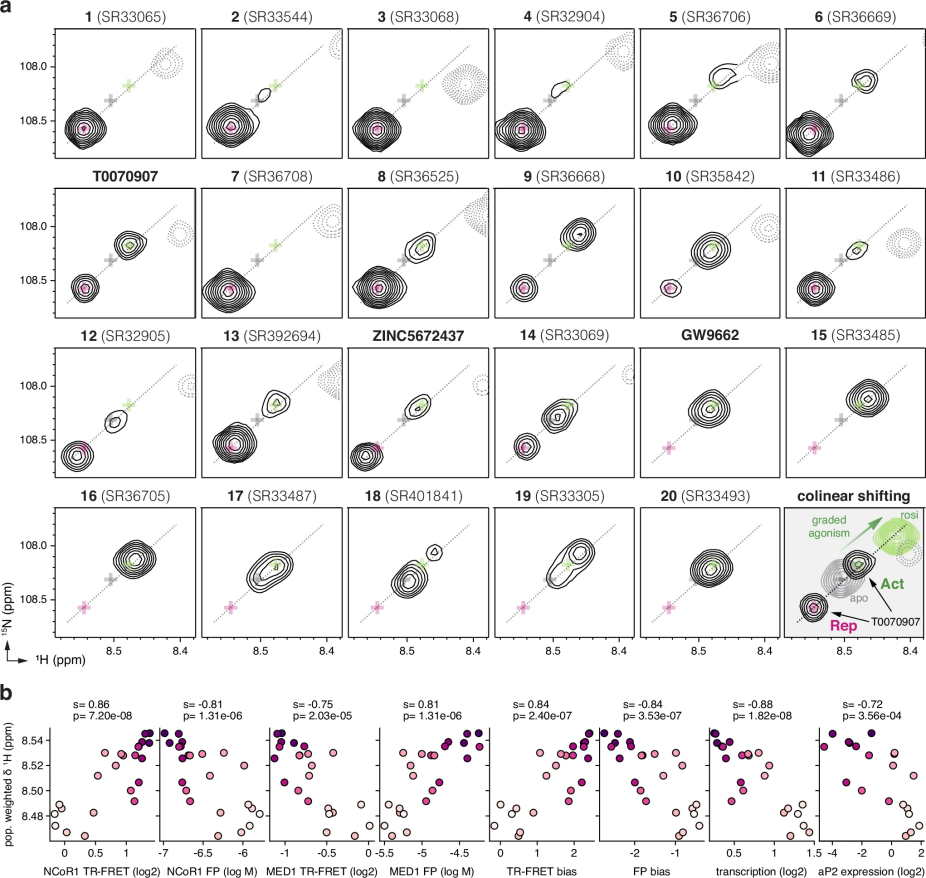
图 6
图6a通过核磁共振波谱展示了不同类型配体对PPARγ构象的影响:
图6b通过定量分析揭示了配体调控机制。研究人员计算了Gly399位点的核磁共振化学位移值,这个参数反映了化合物对蛋白质构象的影响程度。斯皮尔曼相关系数分析表明,NMR数据与生化和细胞实验结果高度相关(相关系数>0.7)。这证实了研究团队的假设:这些化合物是通过调节PPARγ构象的平衡来实现其不同程度的生物学效应。尽管在NMR峰积分等技术细节上存在一些局限性,但整体数据支持配体通过影响蛋白质构象来调控其功能的作用机制。
讨论
本研究通过比较晶体学和核磁共振方法的优劣,强调了研究核受体动态调控机制时需要合适的技术手段。传统的晶体结构虽然能提供高分辨率的静态图像,但无法解释配体如何实现其渐进式调控作用。而核磁共振技术则可以揭示PPARγ配体结合域的动态构象变化,为理解配体调控机制提供了关键信息。研究团队基于T0070907开发的化合物系列具有独特优势:通过简单的化学修饰就能实现从转录抑制到激活的精确调控。这些化合物的作用机制是通过选择性地稳定受体蛋白天然存在的特定构象来实现的。
研究还发现PPARγ反向激动剂存在两种不同的作用模式:选择性稳定模式能够选择性地稳定受体的天然抑制态构象,而竞争性结合模式通过与螺旋12竞争结合位点来实现抑制作用。这项研究不仅建立了评估PPARγ配体活性的分析平台,而且为开发治疗膀胱癌的PPARγ靶向药物提供了重要的理论基础。特别是考虑到目前已有共价结合型PPARγ反向激动剂进入临床试验阶段,本研究提供的构象-活性关系分析方法将对药物开发具有重要的指导意义。
编译|于洲
审稿|王梓旭
参考资料
MacTavish B S, Zhu D, Shang J, et al. Ligand efficacy shifts a nuclear receptor conformational ensemble between transcriptionally active and repressive states[J]. Nature Communications, 2025, 16(1): 2065.
内容中包含的图片若涉及版权问题,请及时与我们联系删除
