

通过根据目标受体的特性直接生成潜在的结合化合物,可以有效缩短早期药物设计阶段。传统上,药物设计很大程度上依赖于高通量筛选(HTS),它缺乏选择要测试的化合物的先验信息。在本论文中,我们将受体的特性整合到深层生成模型框架中,以直接有效地生成高结合化合物。第 1 章至第 4 章提供了药物设计和深度学习方法的背景信息。随后的章节正式介绍我们的工作。第一部分介绍了一种由图神经网络和通用对抗网络组成的设计,用于针对受体特异性的形状受限的小分子药物。该方法为给定受体生成 3D 构象就绪分子,执行支架跳跃和从头配体设计任务。事实证明,与 Enamine REAL 等标准 HTS 数据集相比,形状约束分子生成器在为受体生成高结合分子方面更有效。第二部分介绍了在潜在空间中运行的蒙特卡罗采样方法,以生成蛋白质结合的特异性肽药物。该方法结合了分子动力学模拟的有限反馈迭代,通过计算和实验鉴定了两个蛋白质系统中有效的结合肽药物。随后的改进包括重新设计肽采样器的优化循环,允许更多的反馈迭代并在更短的时间内生产出具有优异结合质量的肽。总之,我们的工作展示了如何将受体的特性纳入深度学习模型来提高早期药物设计过程的效率。

论文题目:Deep Generative Methods For Target Specific Drug Design
作者:Tong Lin
类型:2024年博士论文
学校:Carnegie Mellon University(美国卡内基梅隆大学)
下载链接:
链接: https://pan.baidu.com/s/1EBxdr7AO9CQQMWSShrzq2A?pwd=has4
硕博论文汇总:
链接: https://pan.baidu.com/s/1Gv3R58pgUfHPu4PYFhCSJw?pwd=svp5
药物发现的过程既耗时又昂贵。平均而言,将一种药物推向市场大约需要12年时间和20亿美元的投资[6]。图 1.1 说明了药物开发成本不断上升,从 1970 年代的 1.79 亿飙升至 2010 年代初的 26 亿。药物设计初始阶段临床试验的失败极大地增加了药物开发过程的总体费用。为了降低成本和时间,缩短药物设计周期的持续时间并提高临床前阶段的设计质量至关重要。
自 20 世纪 60 年代以来,机器学习模型在应对这些挑战方面发挥了关键作用,创建了结构-活性关系 (SAR) 模型来根据现有数据预测分子特性。20 世纪 80 年代到 2010 年代,对机器学习模型(包括线性回归和回归树)的广泛研究为提高预测精度做出了贡献。这些模型统称为定量构效关系(QSAR)模型,现已集成到各种生物分子评估软件中[7]。如今,分子和蛋白质的实验和模拟数据非常丰富,与传统的 QSAR 方法相比,基于深度学习的模型已表现出更出色、更快速的特性预测。此外,它们表现出有效生成具有所需特性的分子的能力。
然而,药物设计与分子设计不同,因为许多药物需要对特定受体具有特异性。这一限制限制了深度学习模型的应用,仅针对具有充足数据的受体设计药物。因此,值得探索能够将深度学习模型应用于一般目标特异性药物设计的方法。这一探索旨在加快设计进程,同时确保设计质量。
本论文深入研究了一系列基于深度学习技术的生成建模方法,用于目标特异性药物设计,包括生成肽药物的一级序列和小分子药物的 3D 图结构。靶点特异性药物设计的主要挑战在于靶点结合特异性数据集的可用性有限。
在第一部分中,我们研究了直接在分子结构设计中采用深度学习模型的可行性。生成潜在的小分子药物涉及创建轮廓类似于已识别的蛋白质口袋的分子,并辨别分子和目标蛋白质之间的关键相互作用以确保有效结合。小分子药物设计的优势在于利用丰富的化合物结构数据集来促进基于结构的设计。
论文的后续部分重点讨论肽序列设计,由于肽药物的结构复杂且结合数据稀缺,这是一项更加复杂的任务。虽然肽的结合取决于蛋白质-肽复合物的结构,但目前蛋白质肽相互作用的复杂性以及特定蛋白质的已知结合肽的数量有限,使得基于结构的肽药物设计不切实际。相反,我们转向更可行的生成肽序列的方法,尽管面临与结合数据集稀缺相关的挑战。探索了多种方法,结合模拟和深度学习模型来解决数据稀缺问题,提高生成效率并提高生成的肽的质量。
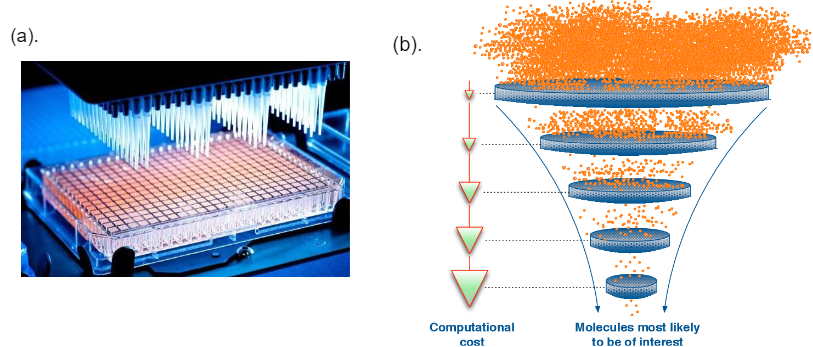 (a) 使用 HTS 进行分子平行筛选 [1]。(b) 分层虚拟筛选 [2]
(a) 使用 HTS 进行分子平行筛选 [1]。(b) 分层虚拟筛选 [2]
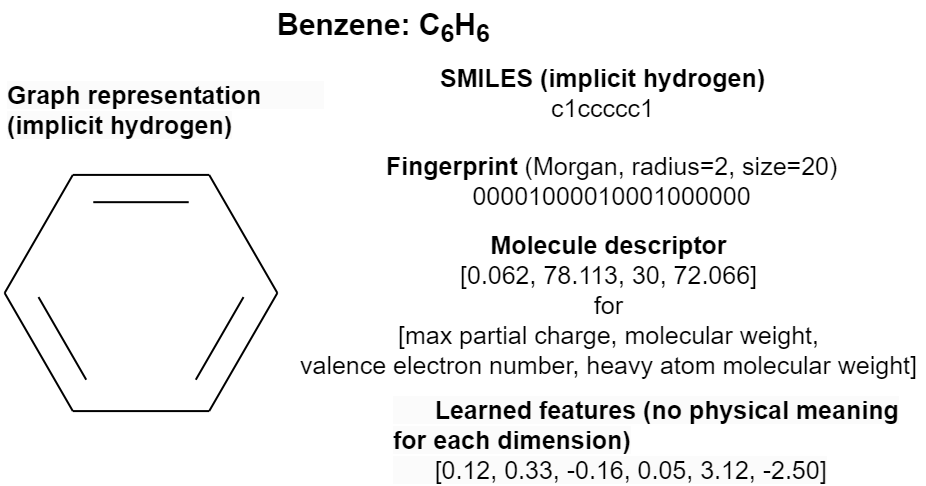 苯的不同表示
苯的不同表示
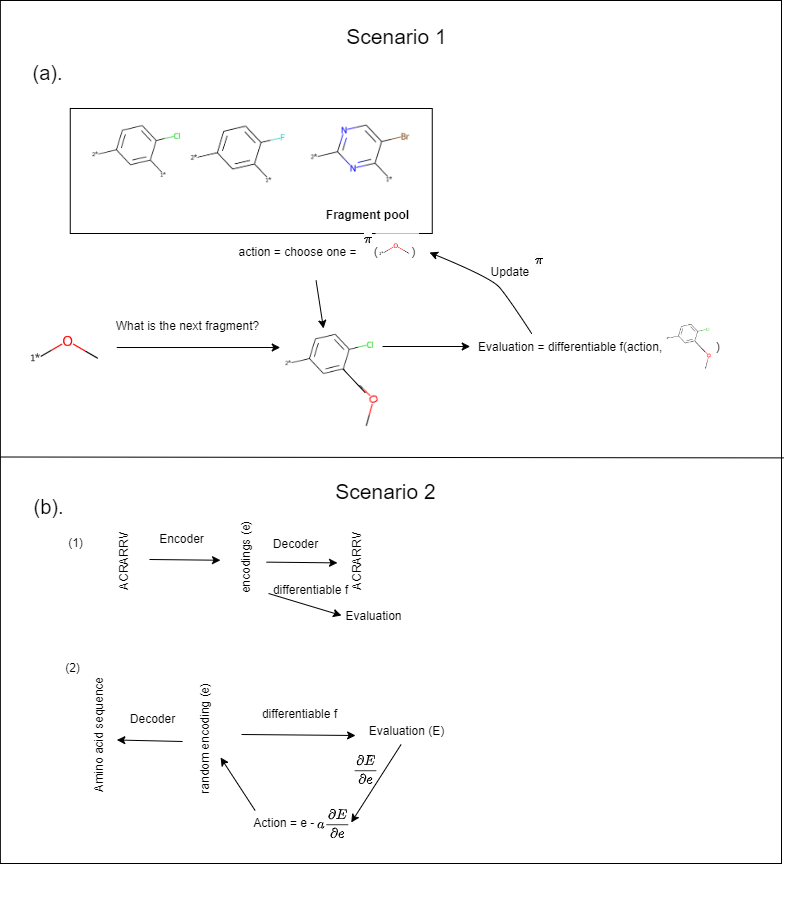 (a) 在每一步中,都需要选择一个片段以添加到池中。该操作由可微策略 π 指导,该策略是在评估先前步骤的操作结果后学习的。(b) 使用自动编码器学习分子表示。连续编码用作分子表示,直接使用可微函数进行评估。因此,只要动作在此表示空间中运行,梯度下降算法就可以用作动作策略。
(a) 在每一步中,都需要选择一个片段以添加到池中。该操作由可微策略 π 指导,该策略是在评估先前步骤的操作结果后学习的。(b) 使用自动编码器学习分子表示。连续编码用作分子表示,直接使用可微函数进行评估。因此,只要动作在此表示空间中运行,梯度下降算法就可以用作动作策略。
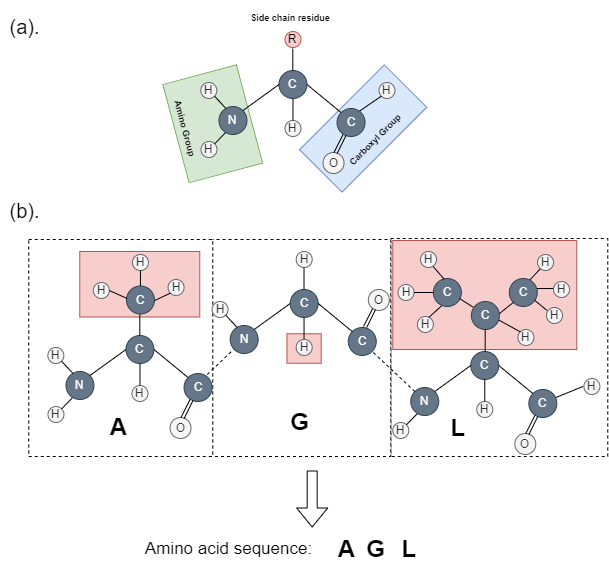 (a) 所有氨基酸均由氨基、羧基和侧链残基组成。不同之处在于侧链残基的形式不同。(b) 大多数蛋白质是由氨基酸串联而成。这种连接通常称为肽键,是由氨基酸基团和羧基之间的吸引力形成的,如两个基团之间的虚线所示。由于串联排列,蛋白质可以由不同的氨基酸、氨基酸序列以连续的方式表示。
(a) 所有氨基酸均由氨基、羧基和侧链残基组成。不同之处在于侧链残基的形式不同。(b) 大多数蛋白质是由氨基酸串联而成。这种连接通常称为肽键,是由氨基酸基团和羧基之间的吸引力形成的,如两个基团之间的虚线所示。由于串联排列,蛋白质可以由不同的氨基酸、氨基酸序列以连续的方式表示。
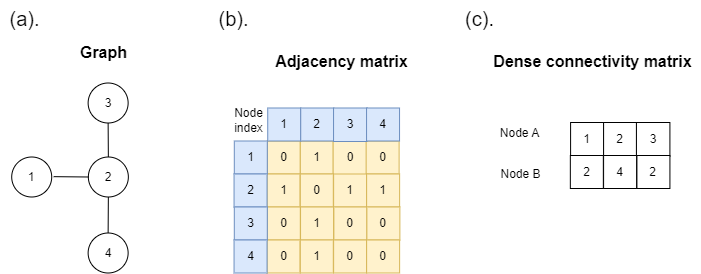 (a) 大小为 4 的示例图。(b) 邻接矩阵有 4 行和 4 列。第a行第b列的值表示节点a和节点b之间的边。因此,行数和列数始终是图形大小。如果值为 1,则连接存在。请注意,邻接矩阵始终是对称的。(c) 连接的密集表示。每列记录由边连接的两个节点。该表示比邻接矩阵小得多并且没有零。请注意,图中的列数与边数相同。
(a) 大小为 4 的示例图。(b) 邻接矩阵有 4 行和 4 列。第a行第b列的值表示节点a和节点b之间的边。因此,行数和列数始终是图形大小。如果值为 1,则连接存在。请注意,邻接矩阵始终是对称的。(c) 连接的密集表示。每列记录由边连接的两个节点。该表示比邻接矩阵小得多并且没有零。请注意,图中的列数与边数相同。
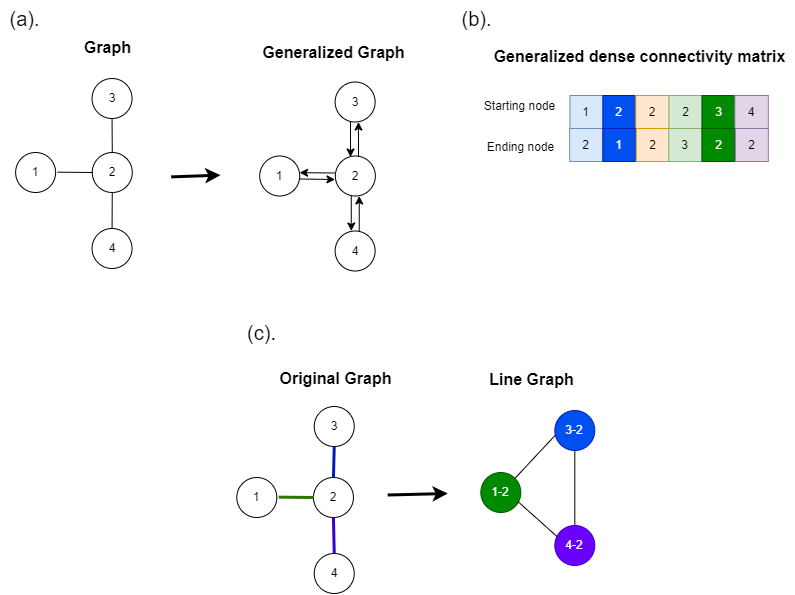
(a) 为了区分边的方向,表示图的更通用的方法是显式地为一条边绘制两个连接,从而可以灵活地区分两个节点之间的信息流的方向。(b) 与图3.3(c)相比,由于需要区分2个节点之间的信息流,列数增加了一倍。具有相似颜色的列具有相同的节点但相反的边。(c) 折线图将原图的边转换为节点。如果两条原始边连接到同一原始节点,则认为它们已连接。该示例显示了这种转换。原始边和变换后的节点之间的对应关系由下式表示颜色。
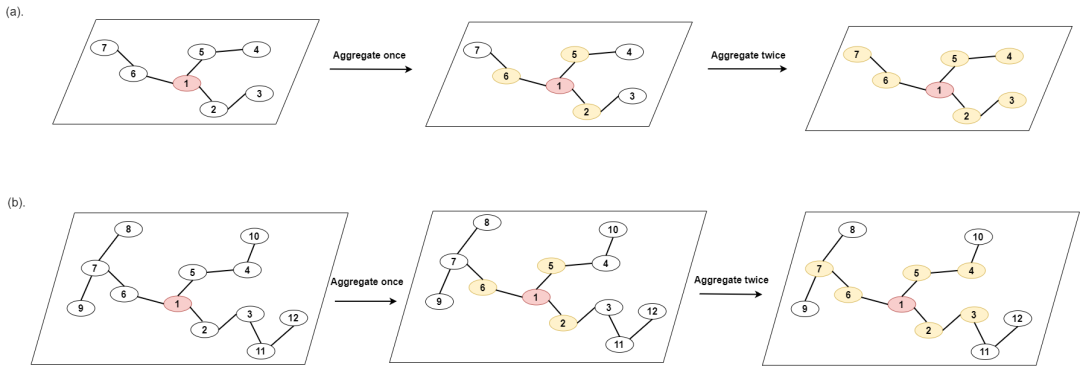 (a) 对于小图,节点 1(黄色节点)经过两层聚合后的感受野可以到达所有节点。(b) 对于大图,节点1经过两层聚合后的感受野只能达到图的一半。在节点级预测任务中,节点 1 的信息量少于 (a) 中对应节点的信息量。
(a) 对于小图,节点 1(黄色节点)经过两层聚合后的感受野可以到达所有节点。(b) 对于大图,节点1经过两层聚合后的感受野只能达到图的一半。在节点级预测任务中,节点 1 的信息量少于 (a) 中对应节点的信息量。
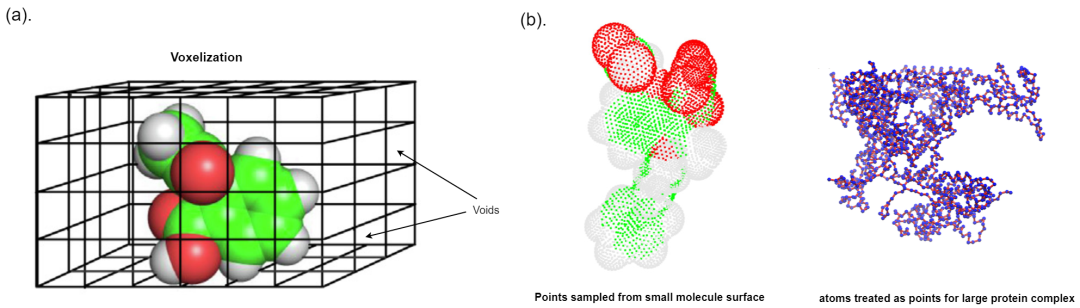 (a) 3D 分子可以体素化成小立方体。然而,由于总体素化空间(由黑框表示)的大小必须在数据集中固定,因此会出现空洞。(b) 有两种使用点云进行分子表示的方法。对于小分子,可以从分子/分子场的表面采样点。对于非常大的分子,其中单个原子与整个结构相比微不足道,原子和键可以被视为点。
(a) 3D 分子可以体素化成小立方体。然而,由于总体素化空间(由黑框表示)的大小必须在数据集中固定,因此会出现空洞。(b) 有两种使用点云进行分子表示的方法。对于小分子,可以从分子/分子场的表面采样点。对于非常大的分子,其中单个原子与整个结构相比微不足道,原子和键可以被视为点。
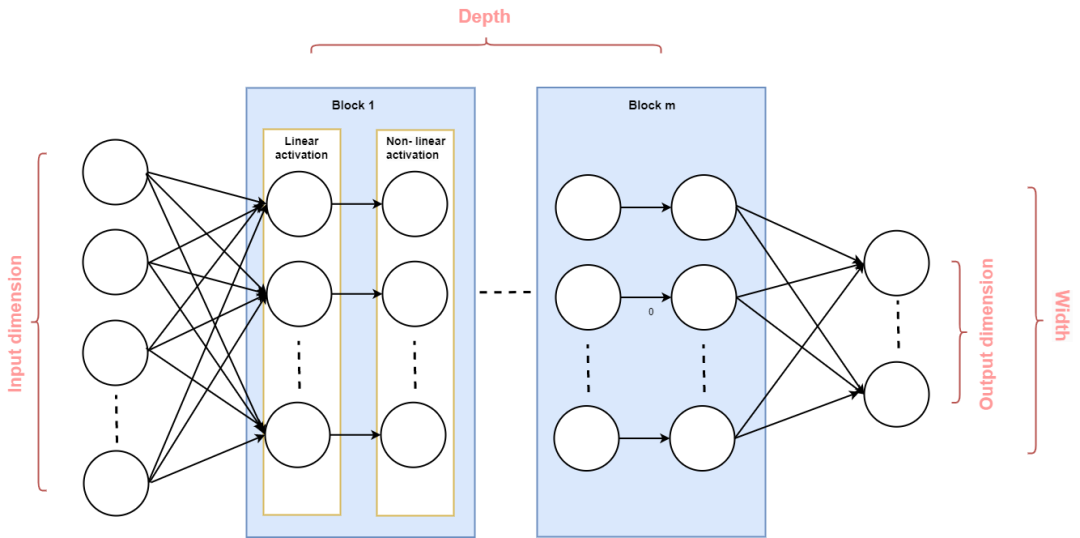
MLP的结构。整个结构是由线性组成的块的重复层和非线性层。块的数量就是 MLP 的深度。线性层(或非线性层)中神经元的数量称为块的宽度。块的最大宽度是 MLP 的宽度。
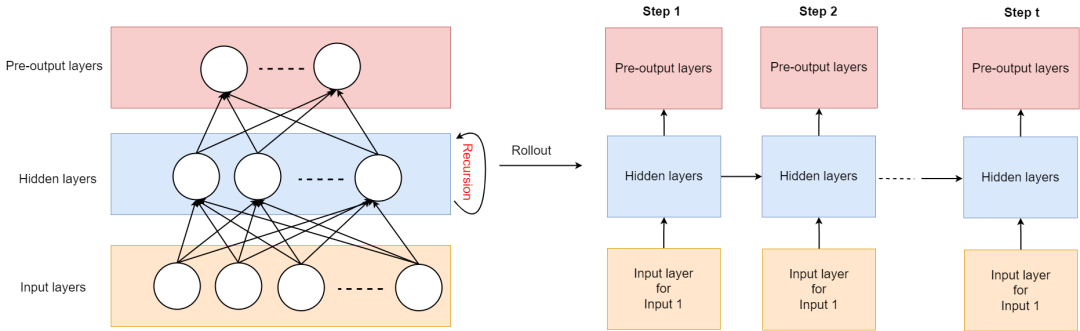 RNN 模型的原始表示和 RNN 结构的推出版本。
RNN 模型的原始表示和 RNN 结构的推出版本。
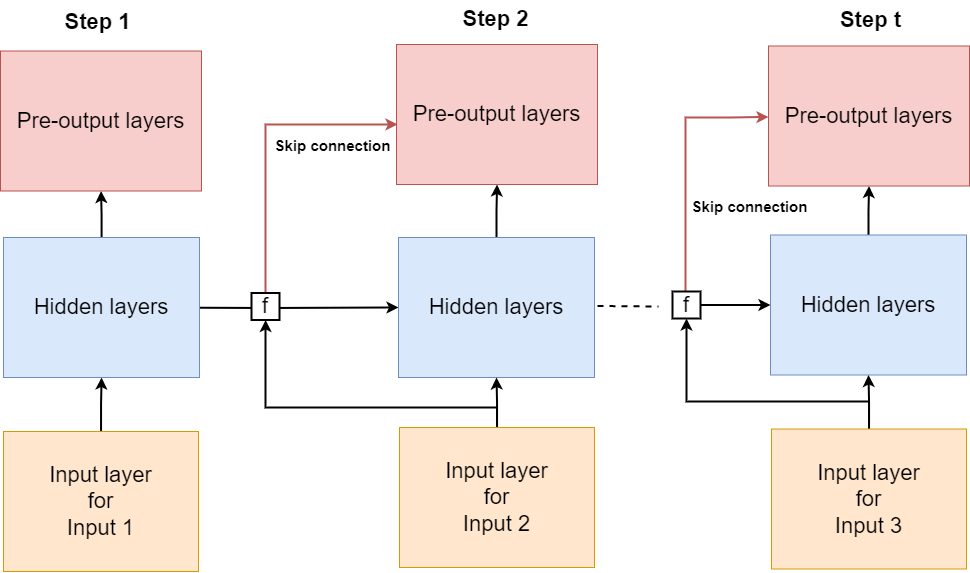
添加了跳过时间连接以促进梯度反向传播问题。对于声称可以解决问题的不同 RNN 模型,主要区别在于连接函数 f。
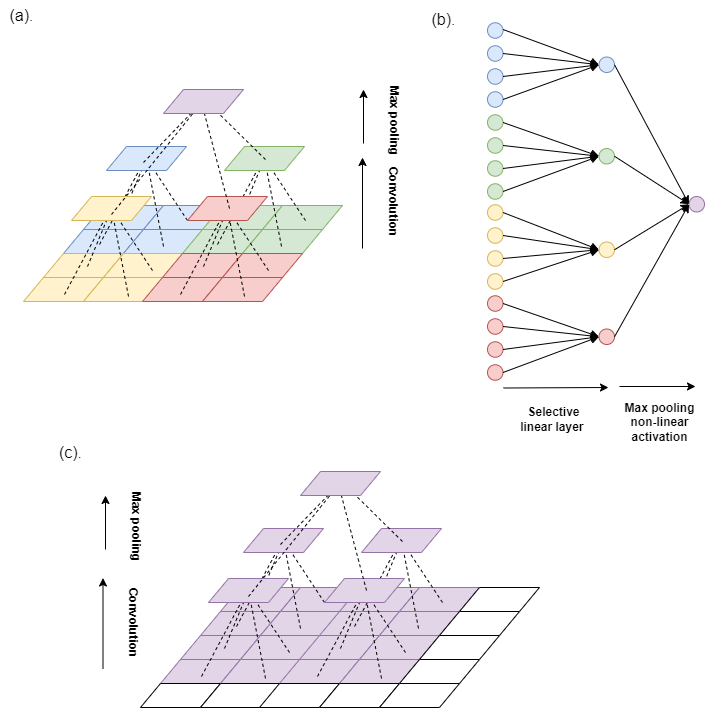 (a) CNN 的常见表示。从下到上的层是输入图像、卷积运算后的输出、最大池化运算后的输出。(b) (a) 的等效图形表示。卷积层本质上是一个具有选择性输入的线性层。最大池化层是一种非激活层。(c) 最大池层之后神经元的感受野。它包含原始图像输入中标记为紫色的所有信息。
(a) CNN 的常见表示。从下到上的层是输入图像、卷积运算后的输出、最大池化运算后的输出。(b) (a) 的等效图形表示。卷积层本质上是一个具有选择性输入的线性层。最大池化层是一种非激活层。(c) 最大池层之后神经元的感受野。它包含原始图像输入中标记为紫色的所有信息。
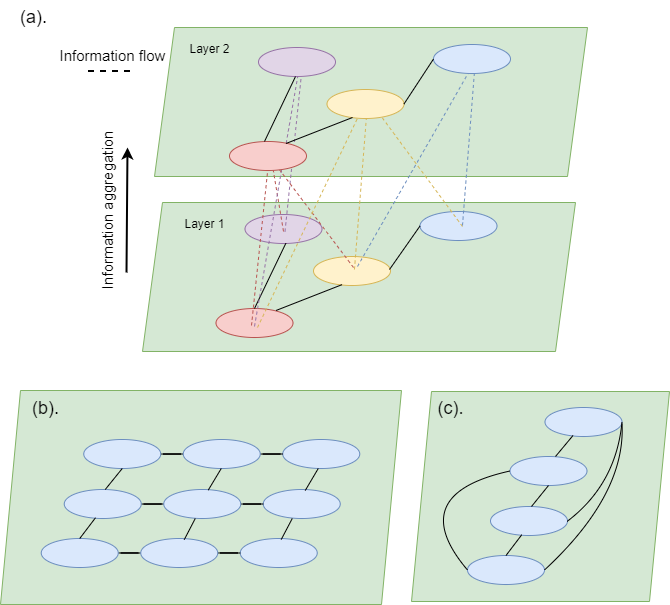 (a) GNN的信息聚合。下一层的节点(神经元)值仅依赖于其自身的值和当前层中相邻节点的值。信息流使用彩色虚线显示。(b) 图像的图形结构表示。该图像是一个平方图。卷积本质上是一种信息聚合操作。(c) MLP 输入的图结构。从图的角度来看,MLP所需的输入是全连接图。那么线性层的操作就相当于GNN的聚合操作。
(a) GNN的信息聚合。下一层的节点(神经元)值仅依赖于其自身的值和当前层中相邻节点的值。信息流使用彩色虚线显示。(b) 图像的图形结构表示。该图像是一个平方图。卷积本质上是一种信息聚合操作。(c) MLP 输入的图结构。从图的角度来看,MLP所需的输入是全连接图。那么线性层的操作就相当于GNN的聚合操作。
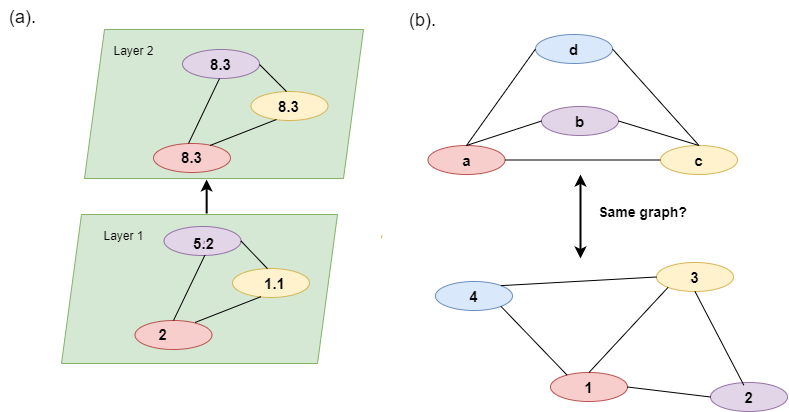 (a) GNN 中的同质性问题。随着 GNN 变得更深,节点值趋于同质。(b) 图同构问题。如果两个图具有相同的拓扑,则它们是同构的。对于一个大图,它的许多子图可以是同构的,这给 GNN 带来了识别问题。
(a) GNN 中的同质性问题。随着 GNN 变得更深,节点值趋于同质。(b) 图同构问题。如果两个图具有相同的拓扑,则它们是同构的。对于一个大图,它的许多子图可以是同构的,这给 GNN 带来了识别问题。
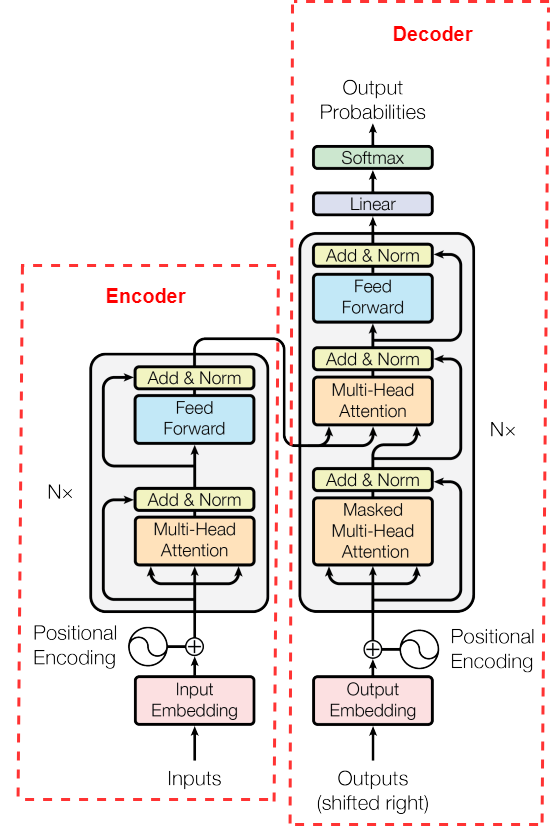
完整的变压器模型。该模型由编码器和解码器组成。两个主要创新是位置编码和多头注意力块。
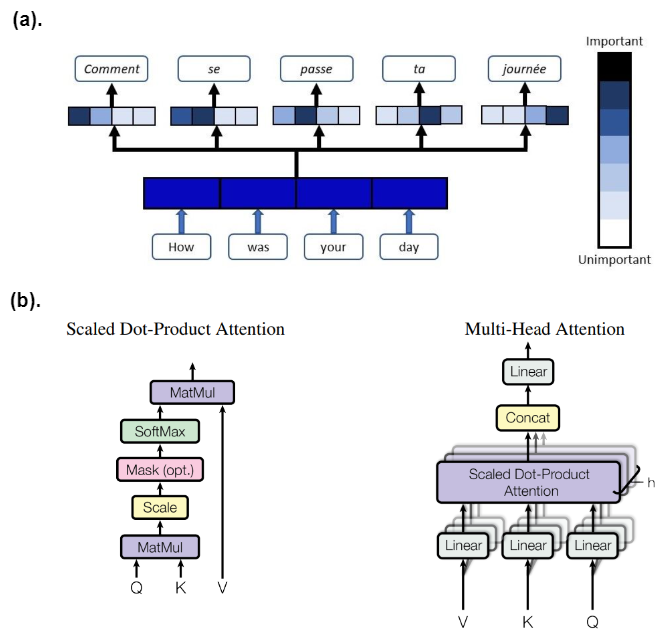
(a)。语言翻译中注意机制的功能说明任务。 (b)。注意力机制的解释。左图是单一注意力机制(一个头)的架构。右图显示了多头注意力机制。它在深度方向上添加具有不同可学习系数的头函数,以通过批量矩阵乘法对输入单词进行批量处理。
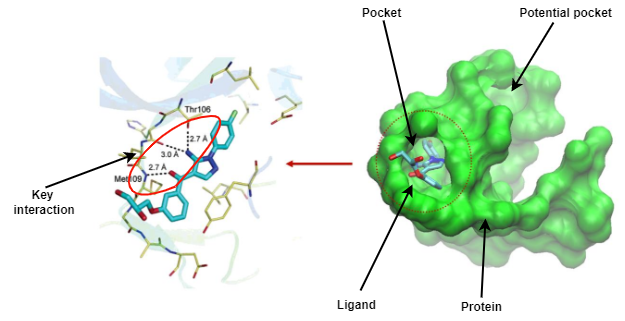 右图显示了蛋白质-配体复合物。左图显示了口袋周围的原子级环境。在右图中,我们可以看到目标蛋白比配体大得多。蛋白质表面有许多空腔和潜在的口袋。在左图中,我们看到蛋白质口袋周围的原子和口袋一侧的配体原子之间存在相互作用。这些相互作用称为关键相互作用,决定了结合的强度。
右图显示了蛋白质-配体复合物。左图显示了口袋周围的原子级环境。在右图中,我们可以看到目标蛋白比配体大得多。蛋白质表面有许多空腔和潜在的口袋。在左图中,我们看到蛋白质口袋周围的原子和口袋一侧的配体原子之间存在相互作用。这些相互作用称为关键相互作用,决定了结合的强度。
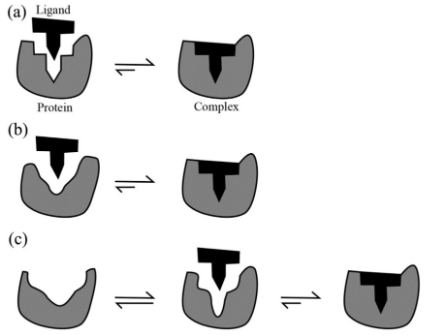 (a) 钥匙和锁的模型。蛋白质和配体都被视为刚体。仅当两种化合物的初始形状匹配时,蛋白质和配体才会结合。(b) 诱导拟合模型。该模型假设蛋白质和配体在结合位点(口袋)都是柔性的。由于配体的存在,口袋的形状发生了变化。这种模型适用于在结合位点诱导变化较小的蛋白质。(c)构象选择模型。这种模型假设整个蛋白质结构在与配体结合之前具有不同的构象。因此,在检查配体-蛋白质结合之前进行构象采样。因此,由于蛋白质构象的变化,口袋可能与初始口袋有很大不同。该模型是三者中对绑定过程描述最准确的模型,同时也是最复杂的模型[3]。
(a) 钥匙和锁的模型。蛋白质和配体都被视为刚体。仅当两种化合物的初始形状匹配时,蛋白质和配体才会结合。(b) 诱导拟合模型。该模型假设蛋白质和配体在结合位点(口袋)都是柔性的。由于配体的存在,口袋的形状发生了变化。这种模型适用于在结合位点诱导变化较小的蛋白质。(c)构象选择模型。这种模型假设整个蛋白质结构在与配体结合之前具有不同的构象。因此,在检查配体-蛋白质结合之前进行构象采样。因此,由于蛋白质构象的变化,口袋可能与初始口袋有很大不同。该模型是三者中对绑定过程描述最准确的模型,同时也是最复杂的模型[3]。
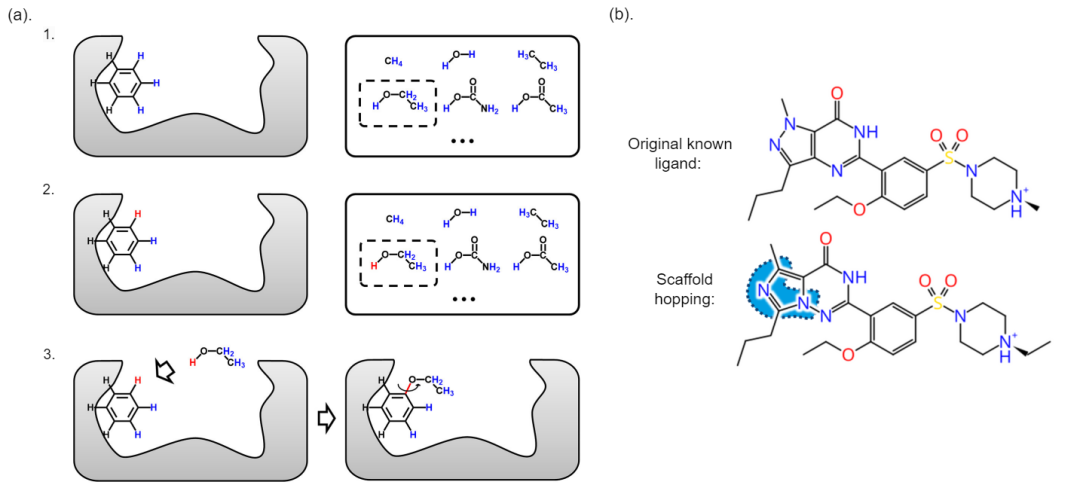 (a) 使用顺序片段添加的从头药物设计。该分子是根据蛋白质口袋的形状构建的。每一步都需要选择片段和链接片段的点。如何制定良好的选择策略是一个活跃的研究课题[4] (b)支架跳跃。它的目的是改变配体的结构但保持其结合特性。支架希望的共同目的是为同一疾病设计新药,同时避免已知配体的专利问题。
(a) 使用顺序片段添加的从头药物设计。该分子是根据蛋白质口袋的形状构建的。每一步都需要选择片段和链接片段的点。如何制定良好的选择策略是一个活跃的研究课题[4] (b)支架跳跃。它的目的是改变配体的结构但保持其结合特性。支架希望的共同目的是为同一疾病设计新药,同时避免已知配体的专利问题。
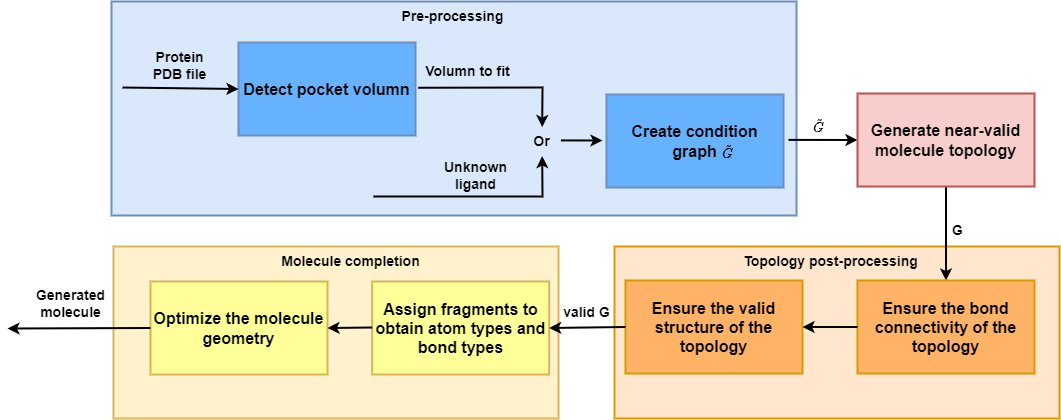 基于口袋的分子生成框架。在预处理步骤中,用作条件 G ̃ 的 3D 图是根据口袋体积或已知配体创建的。然后基于G̃生成接近有效的分子拓扑G。进行后处理以确保生成的 G 是有效的分子拓扑。在最后一步中,执行基于拓扑相似性的基于片段的分配,以分配拓扑的原子类型和键类型。执行基于能量的结构优化以获得最终的分子几何形状。
基于口袋的分子生成框架。在预处理步骤中,用作条件 G ̃ 的 3D 图是根据口袋体积或已知配体创建的。然后基于G̃生成接近有效的分子拓扑G。进行后处理以确保生成的 G 是有效的分子拓扑。在最后一步中,执行基于拓扑相似性的基于片段的分配,以分配拓扑的原子类型和键类型。执行基于能量的结构优化以获得最终的分子几何形状。
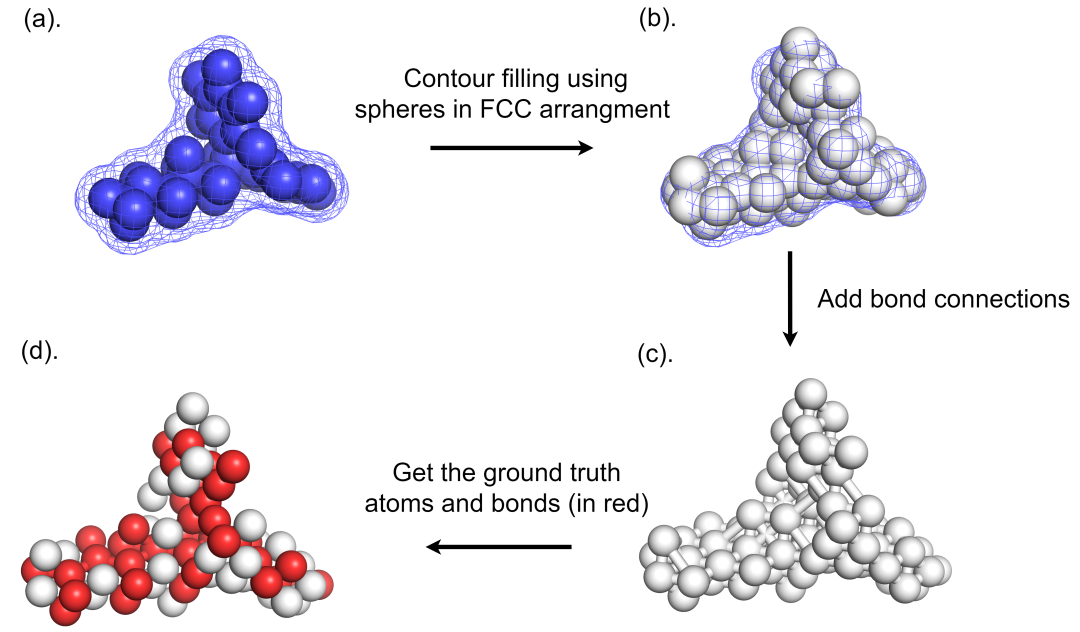 (a) 范德华球中显示的原始分子。蓝色阴影空间是轮廓。(b) 使用晶体结构排列拟合轮廓的范德华球。这给出了变换后的拓扑。(c) 添加键连接后变换后的拓扑。请注意,为了更好的可视化目的,范德华球体的尺寸已减小。(d) 变换拓扑中的真实分子。红色球体和键是最接近原始分子的。而球体和键都是空隙,在拓扑图中用0表示。
(a) 范德华球中显示的原始分子。蓝色阴影空间是轮廓。(b) 使用晶体结构排列拟合轮廓的范德华球。这给出了变换后的拓扑。(c) 添加键连接后变换后的拓扑。请注意,为了更好的可视化目的,范德华球体的尺寸已减小。(d) 变换拓扑中的真实分子。红色球体和键是最接近原始分子的。而球体和键都是空隙,在拓扑图中用0表示。
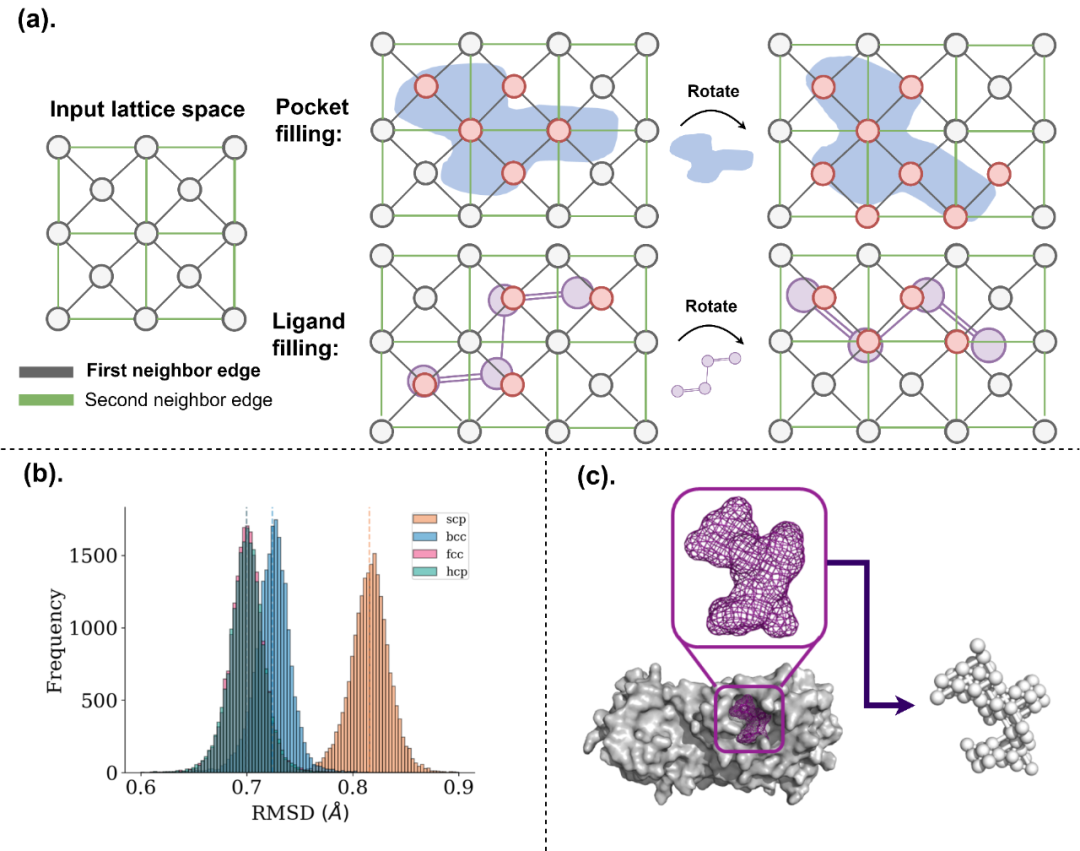 (a) 输入格空间和轮廓填充过程。最左边的图显示了 FCC 排列的输入晶格空间的 2D 示例。中间的图说明了口袋轮廓如何被晶格空间填充。最右边的图说明了已知配体如何被晶格空间填充。(b) 四种不同晶体结构排列的 20640 个分子的填充球体和原始分子之间的 RMSD。(c) 通过填充口袋形成的输入图的示例。
(a) 输入格空间和轮廓填充过程。最左边的图显示了 FCC 排列的输入晶格空间的 2D 示例。中间的图说明了口袋轮廓如何被晶格空间填充。最右边的图说明了已知配体如何被晶格空间填充。(b) 四种不同晶体结构排列的 20640 个分子的填充球体和原始分子之间的 RMSD。(c) 通过填充口袋形成的输入图的示例。
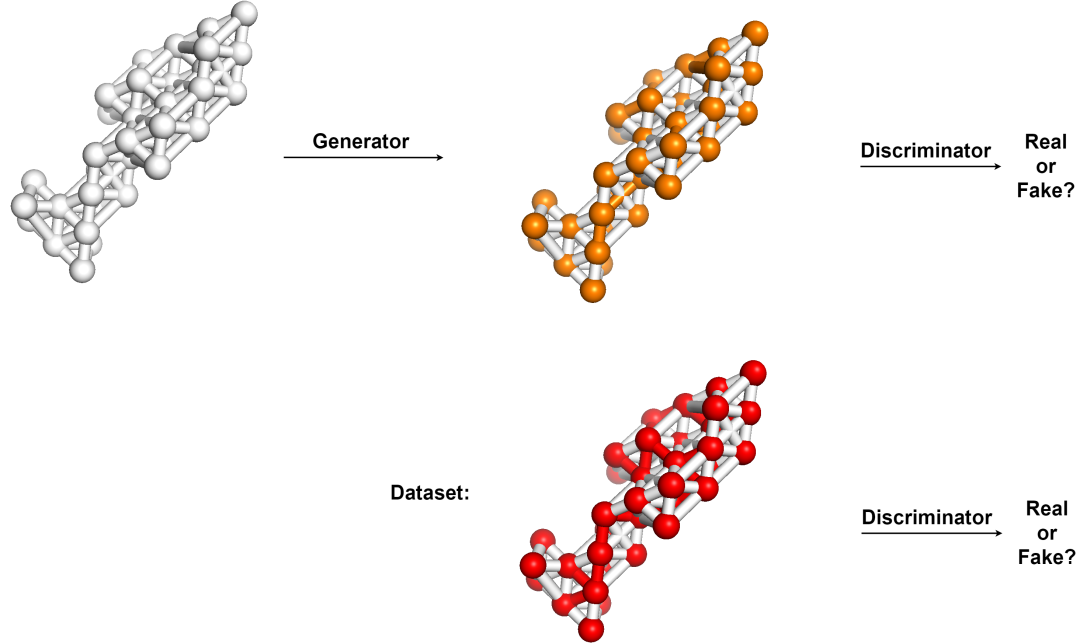 拓扑生成过程。FCC 原子排列由图形结构表示。GAN 框架用于生成近乎真实的分子拓扑。生成是通过识别末端存在哪些节点和边来完成的。那些现有的节点和边以橙色表示。因此,该任务是子图生成任务。
拓扑生成过程。FCC 原子排列由图形结构表示。GAN 框架用于生成近乎真实的分子拓扑。生成是通过识别末端存在哪些节点和边来完成的。那些现有的节点和边以橙色表示。因此,该任务是子图生成任务。
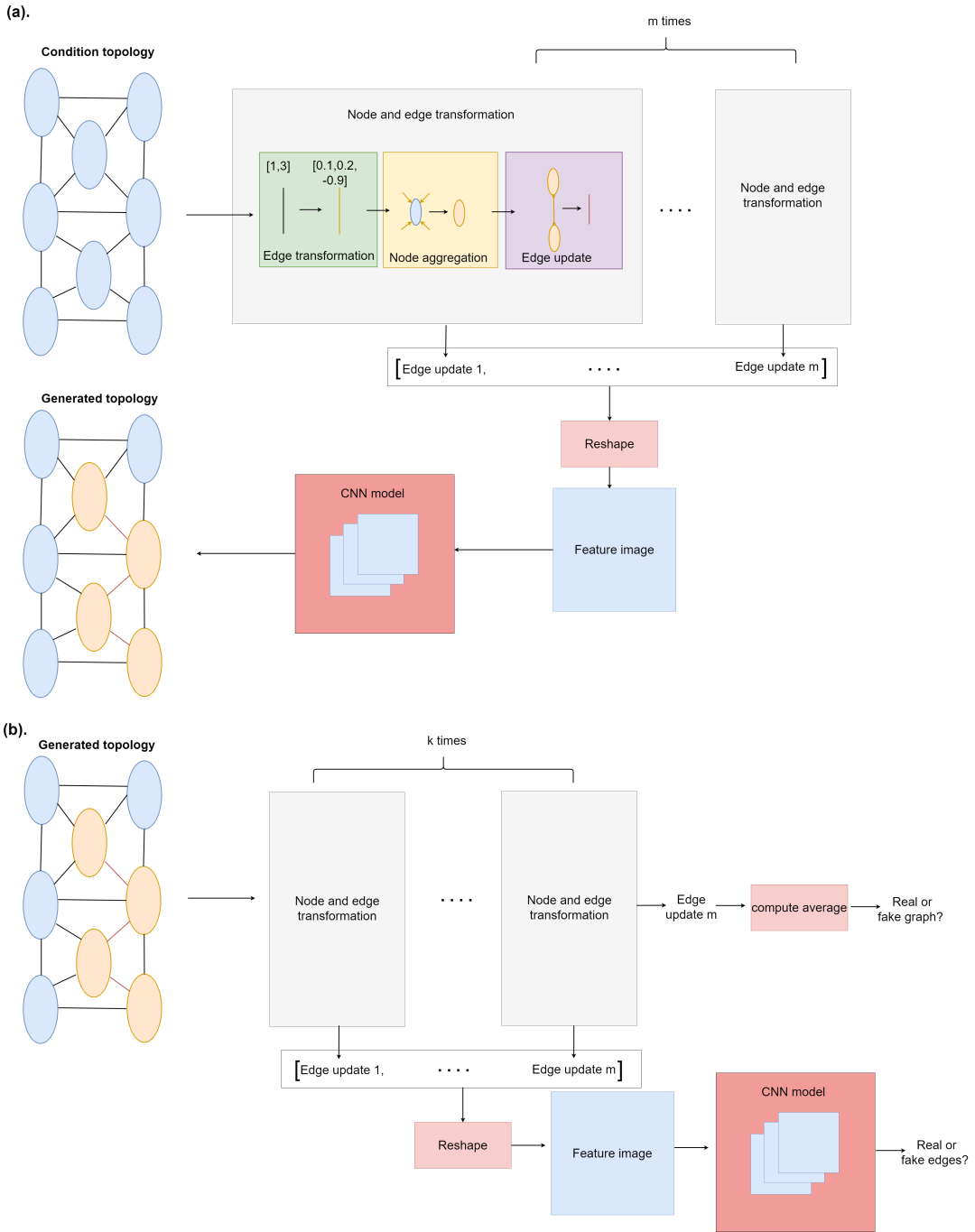 (a) 发电机的模型结构。它由 m 深度的 GNN 模型组成,其中每个图变换块都有 3 个主要组成部分:边变换、节点聚合和边更新。收集每个深度输出的边缘更新,以避免深层结构的节点同质性问题。所有收集到的边缘更新都会被整形并输入 CNN 模型以获得最终生成的拓扑。(b) 判别器的模型结构。判别器有两个识别任务,图识别和边缘识别。图识别控制生成图好坏的全局判断。边缘识别控制局部特征,例如环和边缘连接性。尽管 GNN 通常存在图同构问题,但我们观察到添加这种边缘识别可以提高局部特征生成质量。
(a) 发电机的模型结构。它由 m 深度的 GNN 模型组成,其中每个图变换块都有 3 个主要组成部分:边变换、节点聚合和边更新。收集每个深度输出的边缘更新,以避免深层结构的节点同质性问题。所有收集到的边缘更新都会被整形并输入 CNN 模型以获得最终生成的拓扑。(b) 判别器的模型结构。判别器有两个识别任务,图识别和边缘识别。图识别控制生成图好坏的全局判断。边缘识别控制局部特征,例如环和边缘连接性。尽管 GNN 通常存在图同构问题,但我们观察到添加这种边缘识别可以提高局部特征生成质量。
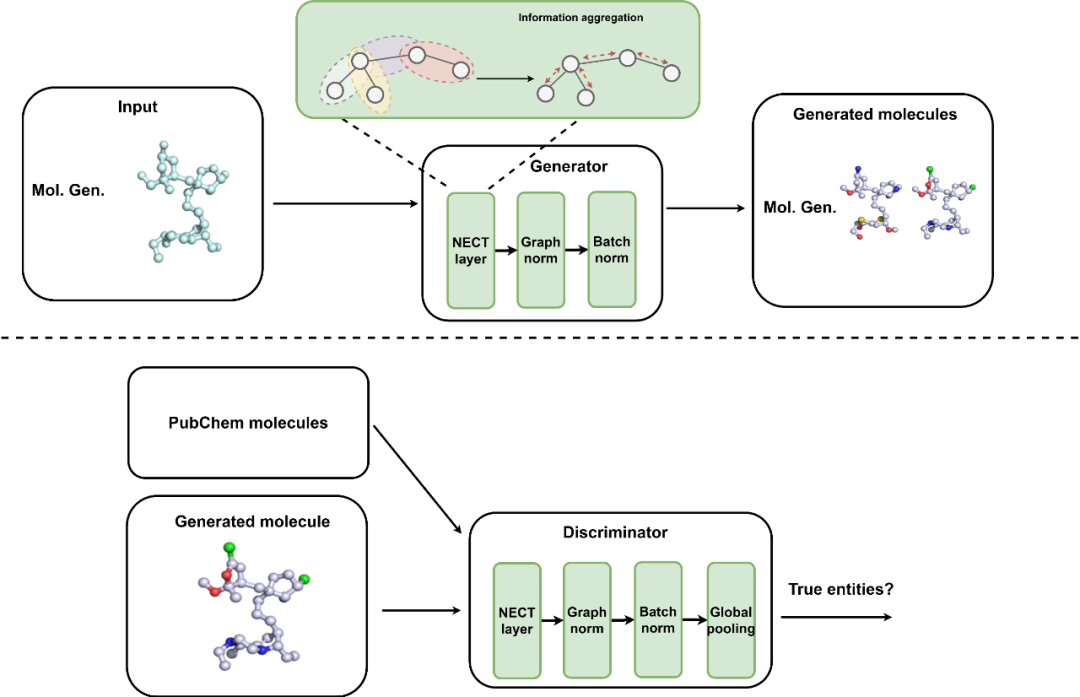 基于 GNN-GAN 的原子键分配模型。上图是发电机。生成器可以采用一种拓扑并对不同的分子进行采样。下图是鉴别器。鉴别器试图判断一个分子是来自 PubChem 数据集还是生成的。
基于 GNN-GAN 的原子键分配模型。上图是发电机。生成器可以采用一种拓扑并对不同的分子进行采样。下图是鉴别器。鉴别器试图判断一个分子是来自 PubChem 数据集还是生成的。
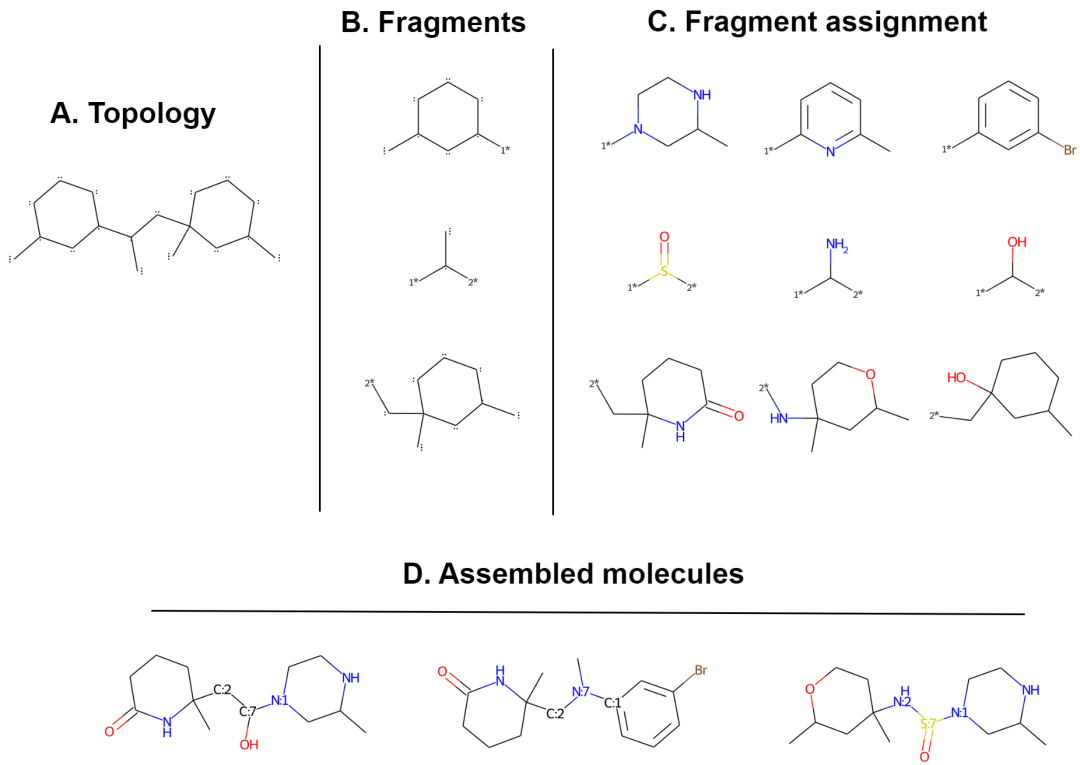 分子片段分配的过程。原始拓扑在不在环上的键上被切断。这里应用了 2 次切割,产生了 3 个片段。每个拓扑片段需要与提供的数据集中现有的分子片段相匹配。然后匹配的分子片段在切割点组装形成新的分子。请注意,最终组装的分子具有与初始生成的拓扑相同的拓扑。
分子片段分配的过程。原始拓扑在不在环上的键上被切断。这里应用了 2 次切割,产生了 3 个片段。每个拓扑片段需要与提供的数据集中现有的分子片段相匹配。然后匹配的分子片段在切割点组装形成新的分子。请注意,最终组装的分子具有与初始生成的拓扑相同的拓扑。
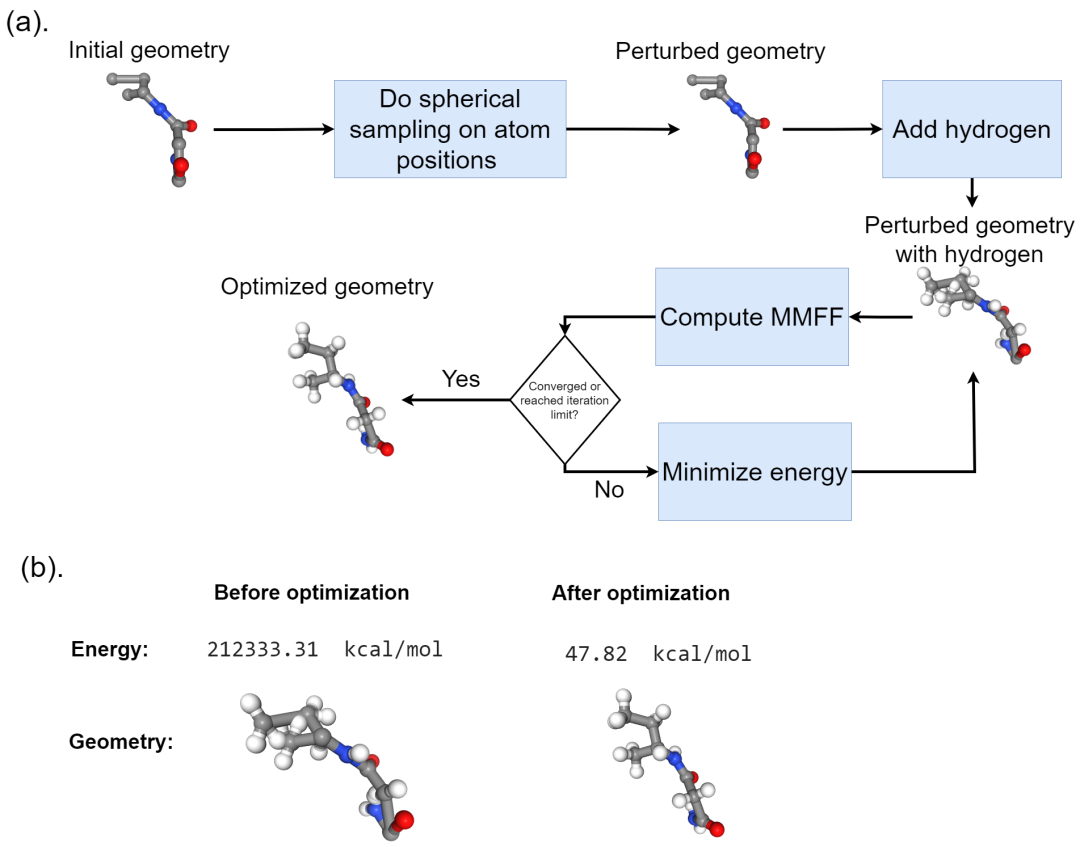 (a) 分子结构优化程序。对每个原子应用球形采样以产生小的扰动。由于我们的原子是 FCC 定位的,一些原子可能会排列在一条线上。这种扰动可以避免优化过程中的数值问题。结构优化时采用MMFF作为能量评估方法。(b) 结构优化前后分子的比较。我们看到优化后分子的总能量显着降低。
(a) 分子结构优化程序。对每个原子应用球形采样以产生小的扰动。由于我们的原子是 FCC 定位的,一些原子可能会排列在一条线上。这种扰动可以避免优化过程中的数值问题。结构优化时采用MMFF作为能量评估方法。(b) 结构优化前后分子的比较。我们看到优化后分子的总能量显着降低。
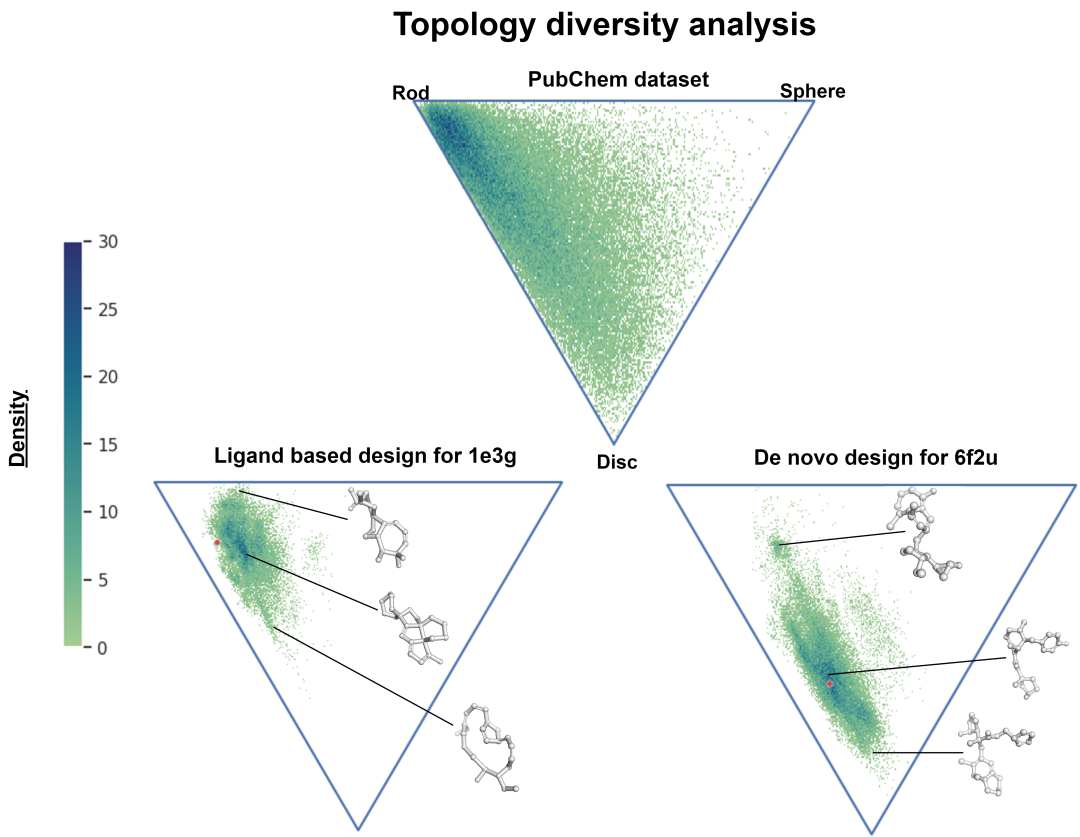
生成分子的拓扑结构分析。三种特征形状(棒、圆盘和球体)位于 NPR 三角形图的拐角处。上图是 PubChem 数据集的拓扑形状分布。左下角是受体 1e3g 的基于配体的分子生成。右下角是受体 6f2u 的基于口袋的分子生成(假设配体形状未知)。
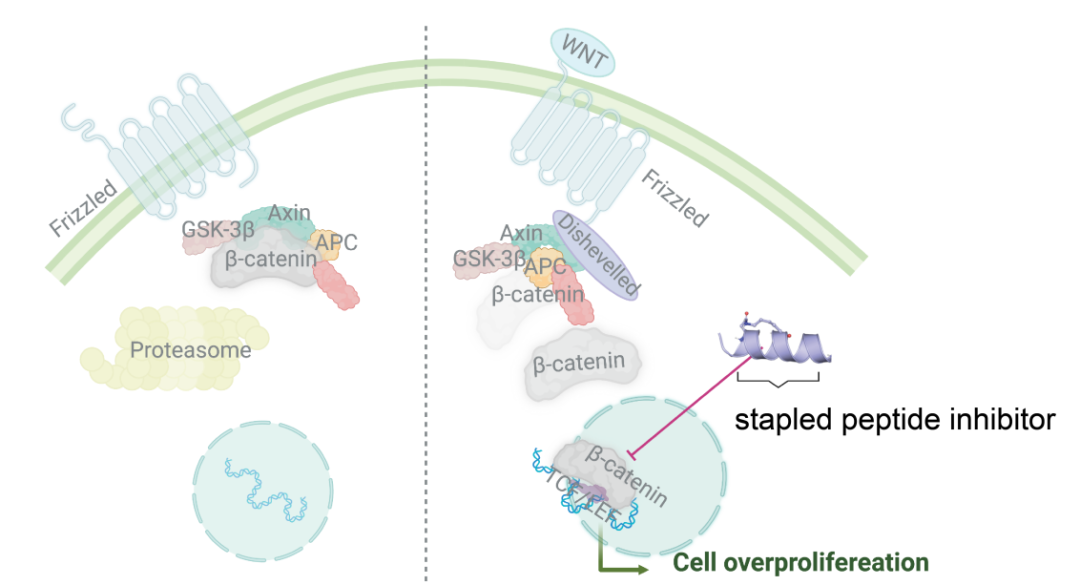 β-连环蛋白与细胞过度增殖之间关系的说明。β-连环蛋白最初存在于细胞中,如左图所示。然而,它有机会进入细胞核与基因转录相关的蛋白质相互作用并导致细胞过度增殖。拥有与 β-连环蛋白(肽抑制剂)结合的肽可以有效减少 β-连环蛋白与这些蛋白质的相互作用。
β-连环蛋白与细胞过度增殖之间关系的说明。β-连环蛋白最初存在于细胞中,如左图所示。然而,它有机会进入细胞核与基因转录相关的蛋白质相互作用并导致细胞过度增殖。拥有与 β-连环蛋白(肽抑制剂)结合的肽可以有效减少 β-连环蛋白与这些蛋白质的相互作用。
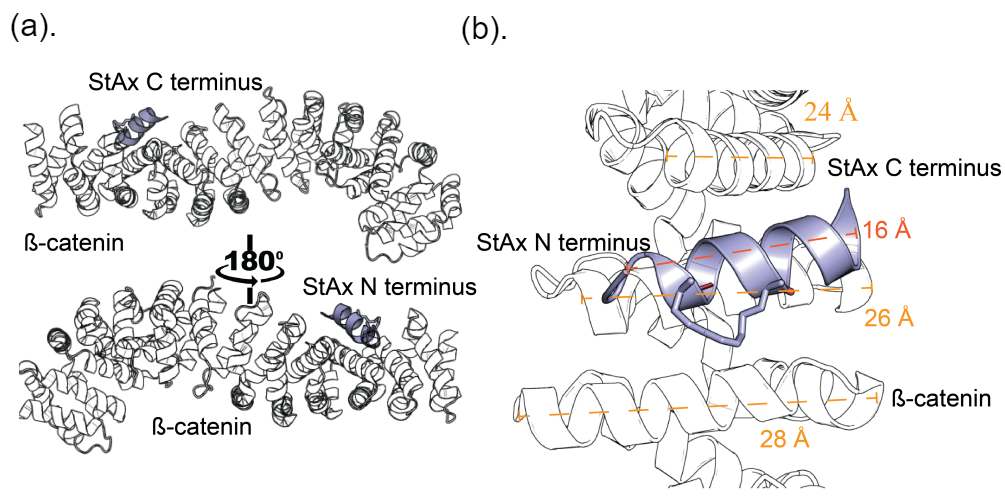 (a) 从肽的 Nor C 末端观察与 β-连环蛋白结合的钉合肽 StAX-35R 的晶体结构 (PDBid:4DJS)。(b) 钉合肽的螺旋长度与 β-连环蛋白的三个相互作用螺旋的比较。
(a) 从肽的 Nor C 末端观察与 β-连环蛋白结合的钉合肽 StAX-35R 的晶体结构 (PDBid:4DJS)。(b) 钉合肽的螺旋长度与 β-连环蛋白的三个相互作用螺旋的比较。
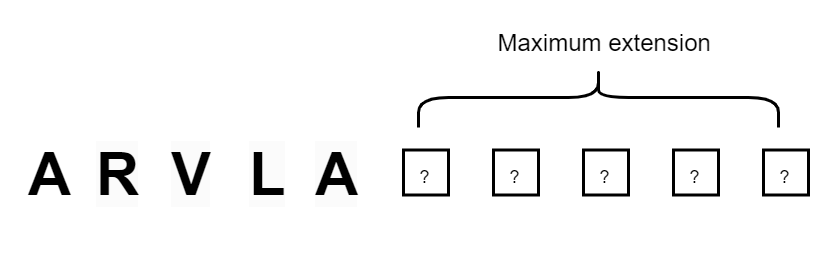
肽延伸任务的问题设置图示
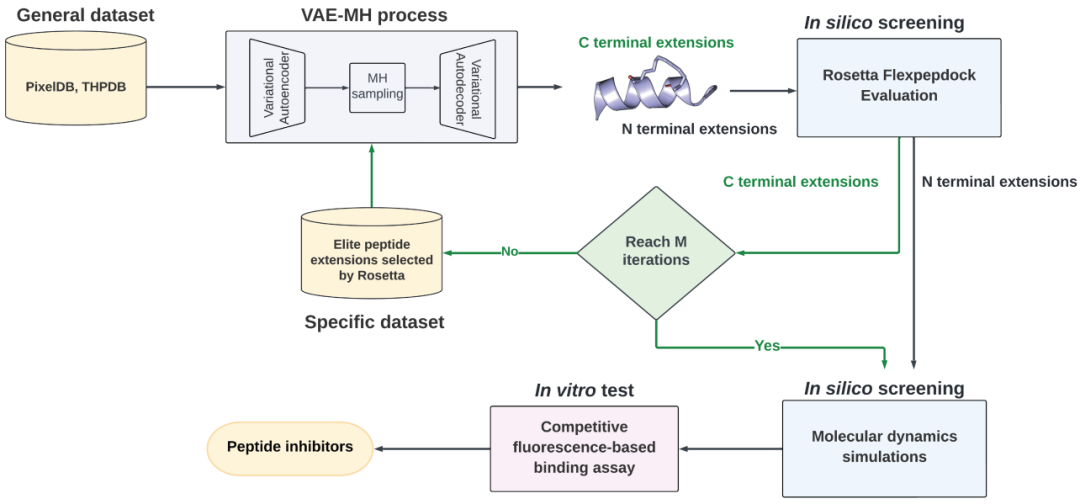 我们的 MD 整合肽设计的整体工作流程
我们的 MD 整合肽设计的整体工作流程
 我们的 MD 整合肽设计的整体工作流程
我们的 MD 整合肽设计的整体工作流程
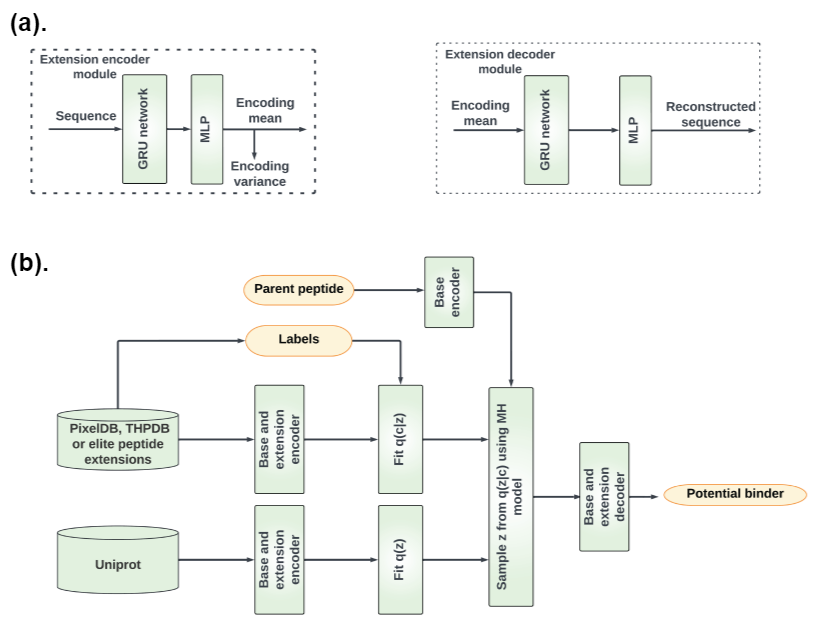 (a)用于编码的GRU-VAE模型。该模型以肽序列嵌入作为输入。它使用 GRU 网络来处理嵌入,并使用 MLP 网络来输出编码均值和方差。解码器使用相同的结构。(b) MH 采样过程。MH 采样框架采用三个输入:肽碱基编码、肽质量判别函数 q(c|z) 和肽序列分布 q(z)。然后,它使用三个输入执行 MH 采样,以对良好的肽延伸进行采样。
(a)用于编码的GRU-VAE模型。该模型以肽序列嵌入作为输入。它使用 GRU 网络来处理嵌入,并使用 MLP 网络来输出编码均值和方差。解码器使用相同的结构。(b) MH 采样过程。MH 采样框架采用三个输入:肽碱基编码、肽质量判别函数 q(c|z) 和肽序列分布 q(z)。然后,它使用三个输入执行 MH 采样,以对良好的肽延伸进行采样。
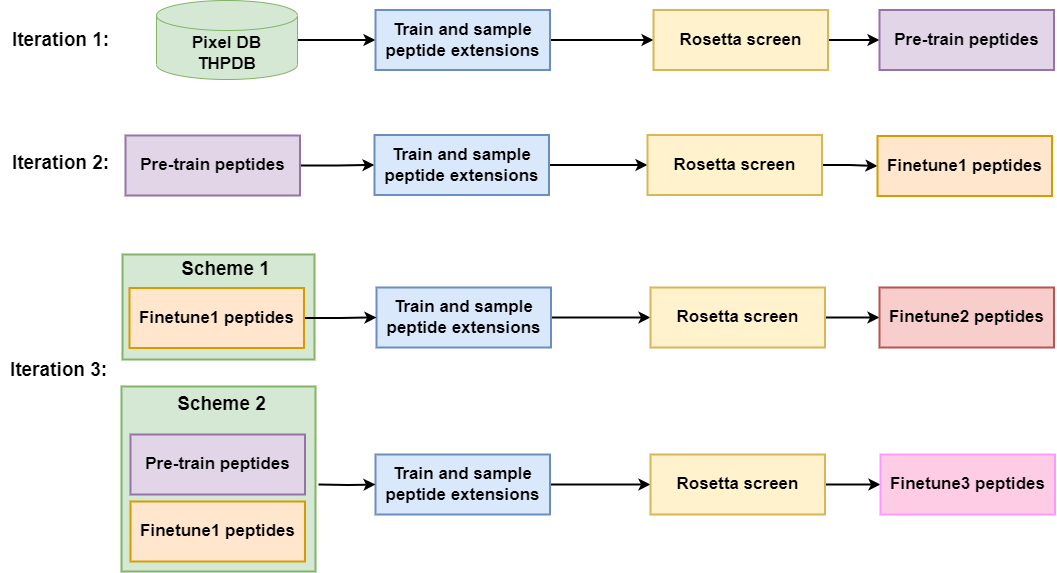 说明如何在三个迭代中使用训练数据集
说明如何在三个迭代中使用训练数据集
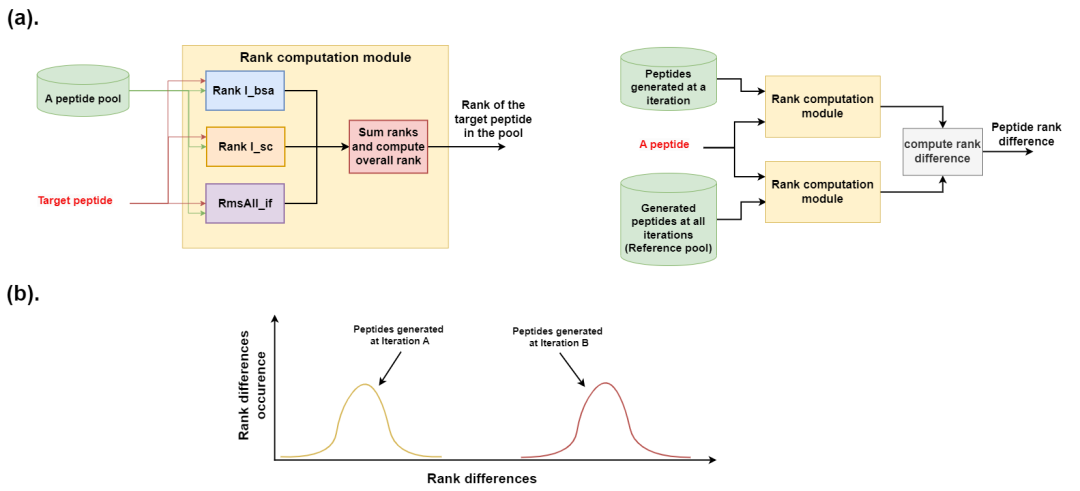
(a) 说明如何计算排名差异。池中肽的排名为通过对每个单独指标的排名总和进行排名来计算,如左图所示。排名差异的计算方式为肽在其生成迭代中的排名与其在所有迭代生成的所有肽中的排名之间的差异,如右图所示。因此,所有迭代生成的所有肽可以被视为当前迭代池中肽的参考池。 (b) 等级差异含义说明。我们可以计算迭代中生成的所有肽的等级差异分布。然后,因为我们有相同的参考池,所以我们可以通过比较两次迭代中肽段的等级差异来比较两次迭代的生成质量。分布向右移动的迭代表明生成的肽更好。因此,我们可以绘制分布来显示迭代的丰富程度。
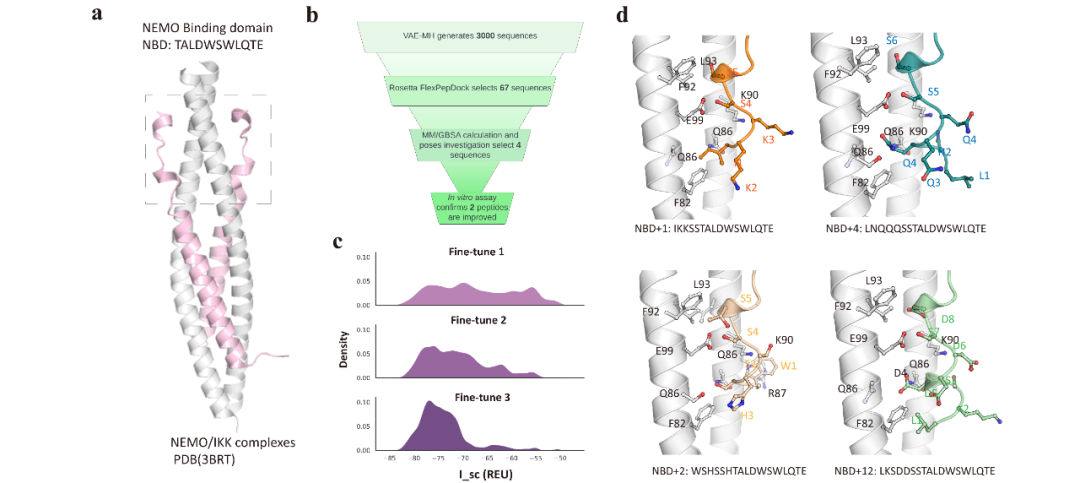
VAE-MH 工作流程在 NEMO/IKK 中的应用。a) NEMO/IKK 复合物的晶体结构(PDBid:3RBT[5])。b) 说明肽序列选择的工作流程。c) 整个微调周期中界面能量(REU)的分布。d) 四种测试肽的肽-NEMO 结合姿势的可视化。NEMO 螺旋以灰色表示,肽抑制剂以不同颜色表示。
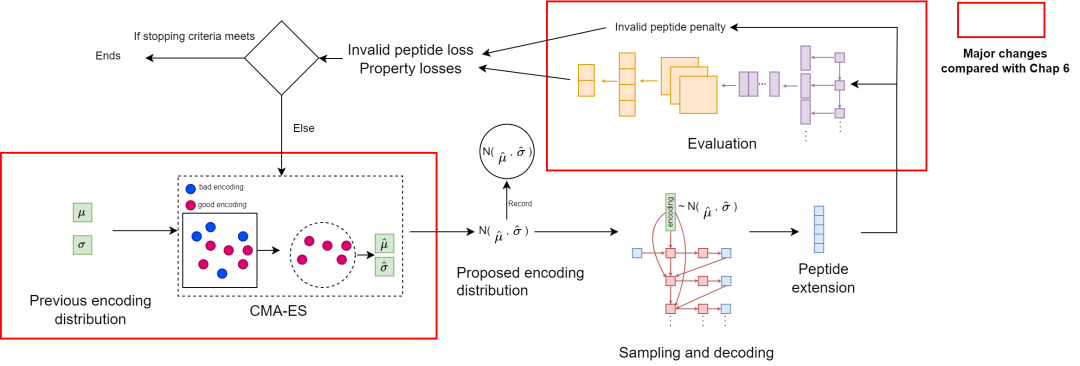 框架概述。MH 采样被 CMA-ES 取代。Rosetta 对接被替代模型取代。同时,记录采样的轨迹,即高斯的 µˆ 和 σˆ,以便以后可以收集好的高斯。
框架概述。MH 采样被 CMA-ES 取代。Rosetta 对接被替代模型取代。同时,记录采样的轨迹,即高斯的 µˆ 和 σˆ,以便以后可以收集好的高斯。
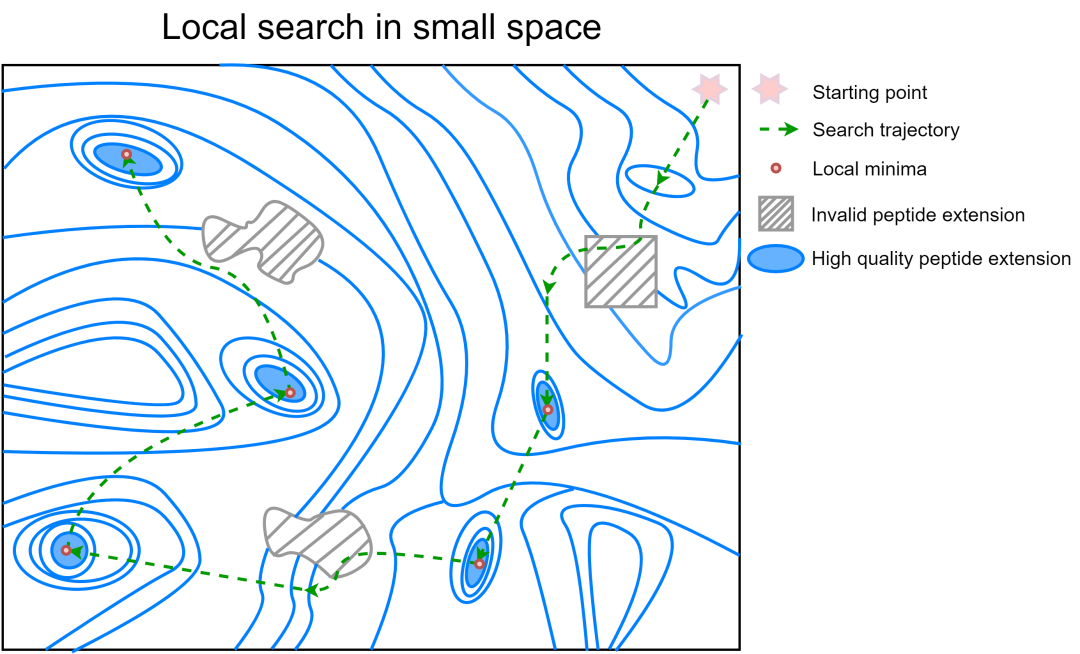
新方法的图示。绿色虚线是Gaussian的优化轨迹用于对肽延伸进行采样的分布。该轨迹可能会穿过几个具有良好肽延伸的小区域。我们记录穿过这些区域的高斯分布。随后,我们使用这些高斯函数对肽延伸进行采样。它避免了整个空间中的样品肽。
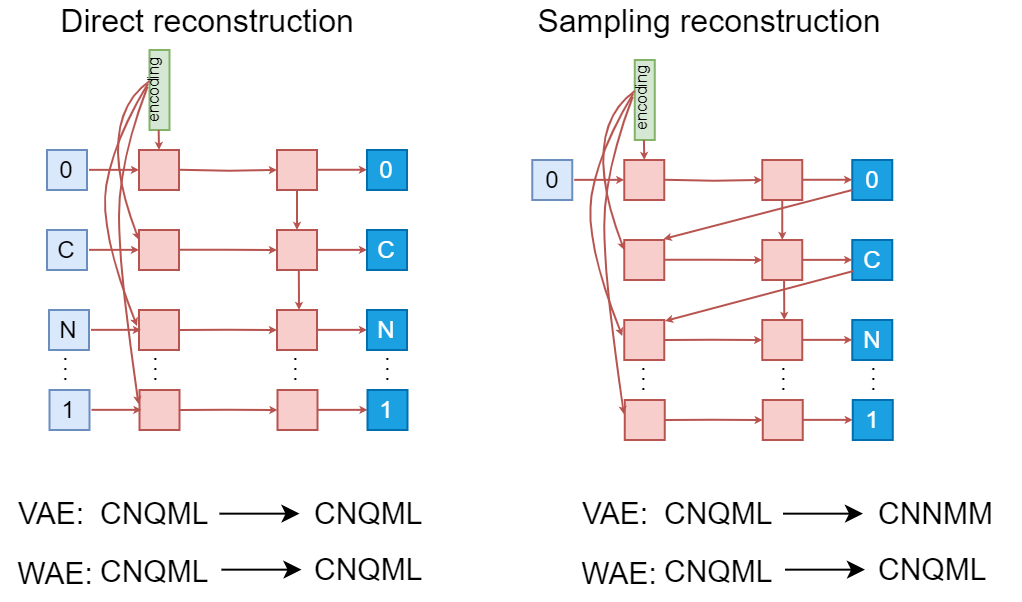 直接重建和采样重建的图示。直接重建输入真实氨基酸和编码以解码下一个氨基酸。采样重建使用预测的氨基酸和编码来解码下一个氨基酸。
直接重建和采样重建的图示。直接重建输入真实氨基酸和编码以解码下一个氨基酸。采样重建使用预测的氨基酸和编码来解码下一个氨基酸。
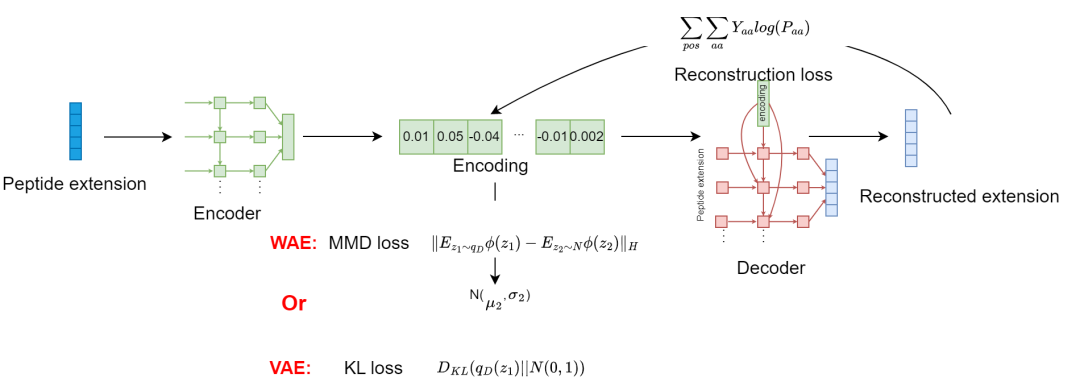 VAE和WAE之间的区别。VAE 和 WAE 之间的主要区别在于它们如何调节编码分布。对于 VAE,编码被调节为单位高斯。对于 WAE,分布被正则化为希尔伯特空间中的高斯分布
VAE和WAE之间的区别。VAE 和 WAE 之间的主要区别在于它们如何调节编码分布。对于 VAE,编码被调节为单位高斯。对于 WAE,分布被正则化为希尔伯特空间中的高斯分布
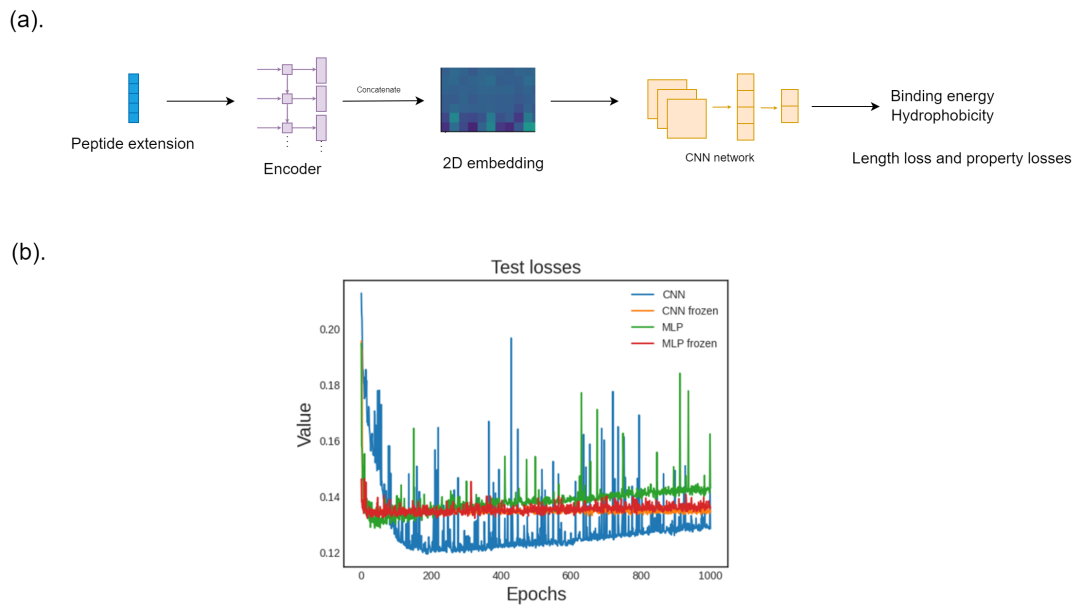 (a) 替代模型结构。(b) 不同代理模型结构的预测损失。
(a) 替代模型结构。(b) 不同代理模型结构的预测损失。
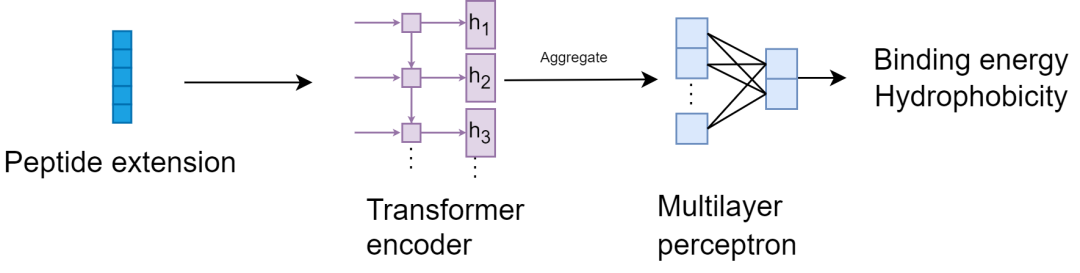 ESM 变压器模型用作替代属性预测模型。
ESM 变压器模型用作替代属性预测模型。







