index_new5.html
../../../zaker_core/zaker_tpl_static/wap/tpl_guoji1.html
![]()
复旦大学王满宁教授团队提出了ProteinF3S框架,旨在通过融合蛋白质序列、结构和表面信息,提升酶功能预测的准确性。该框架创新性地提出了一种蛋白质结构和表面之间的多尺度双向融合策略,使表面和结构编码器能够相互作用,提取更强大的特征。同时,该框架还结合了序列特征,实现了序列、结构和表面信息的有效融合。实验结果表明,ProteinF3S在酶反应分类任务中超越了现有方法,证明了融合不同蛋白质模态信息以及采用有效融合策略的重要性。该研究为蛋白质功能预测提供了新思路,并有望扩展到更多蛋白质任务中。
🧬 ProteinF3S框架融合了蛋白质序列、结构和表面三种模态的信息,旨在提升酶功能预测的准确性,核心在于利用蛋白质不同形态的互补信息。
🧩 提出了多尺度双向融合策略,用于整合蛋白质结构和表面信息。结构和表面编码器通过层次特征的相互作用,提取更强大的特征,有效提升了模型性能。
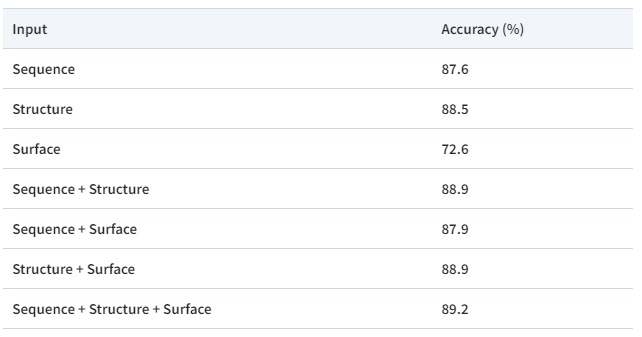
🧪 通过消融实验验证了模型设计的有效性。结果表明,单独使用序列、结构或表面信息,以及两两组合,都比单一模态表现更好。融合所有三种模态信息,可以获得最佳性能。
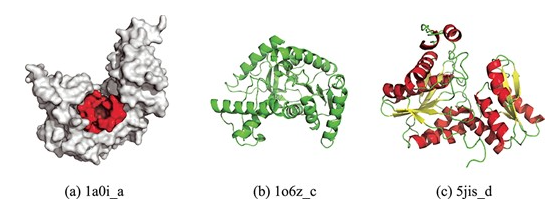
🔬 案例分析揭示了不同蛋白质表示形式的优缺点。表面特征不足以捕捉活性位点的关键属性,而复杂的结构可能会引入噪声。序列信息简洁,但无法直接反映蛋白质的折叠和空间结构。
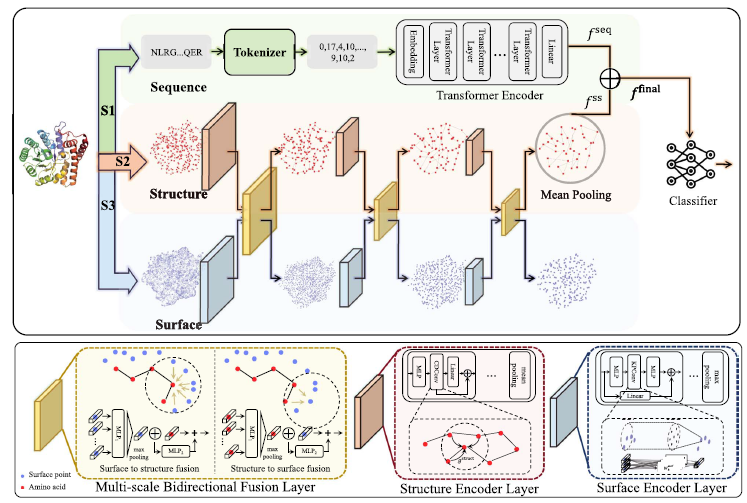
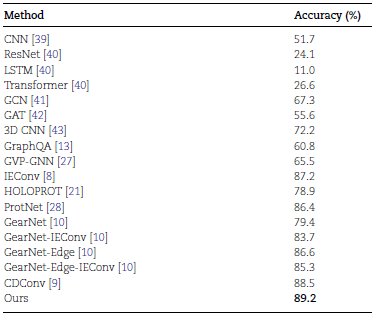
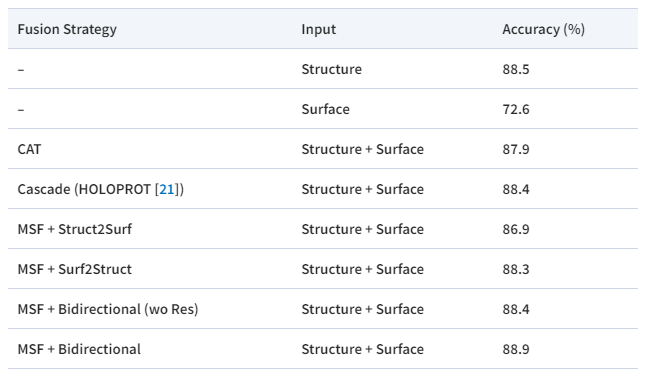
蛋白质可以用不同的数据形式表示,包括序列、结构和表面。与序列和结构信息相比,蛋白质表面信息在此前的研究中相对较少被采用。然而,由于蛋白质的表面直接参与到与其他分子的相互作用,表面信息能在原子水平上提供更清晰的局部特性建模,有效增强序列和结构建模的性能。2025年1月3日,复旦大学王满宁教授团队在Briefings in Bioinformatics上发表文章ProteinF3S: boosting enzyme function prediction by fusing protein sequence, structure, and surface。作者提出了一个新的框架ProteinF3S,该框架融合了蛋白质序列、结构和表面(fusion of sequence, structure and surface)的互补信息。为了实现更有效的融合,作者提出了一种蛋白质结构和表面之间的多尺度双向融合策略,其中表面编码器和结构编码器的层次特征双向相互作用,然后将序列特征与包含结构和表面信息的特征进行串联,从而实现进一步的融合,从而获得更好的性能。实验证明了DMFF-DTA的性能超越了现有方法。如图1所示,ProteinF3S将PDB文件形式的原始蛋白质作为输入,并将其转换为结构、表面和序列。这三种形式的数据分别输入到三个专门的编码器。由于结构编码器和表面编码器都遵循分层结构,因此作者在结构编码器和表面编码器之间采用了多尺度双向融合策略。通过多尺度双向融合,结构和表面的互补信息可以不断丰富两个编码器提取的特征,从而在更高层次上生成更强大的特征。由于序列编码器采用的是全局特征交互而不是层次化的方式,对序列进行截断,使得序列难以与结构或表面对齐。因此,作者连接序列特征与先前多尺度双向融合得到的高级特征以进行融合,得到用于酶反应分类等任务的最终特征表示。将特征表示输入到分类器中,即多层感知器,最终输出预测结果。为了有效地提取不同形式蛋白质的特征表示,作者分别使用了三种专门的蛋白质结构、表面和序列编码器。结构编码器和表面编码器采用分层方式,它们彼此是对称的。序列编码器遵循Transformer架构,将氨基酸序列作为输入并提取序列的全局表示。首先,作者使用CDConv作为结构编码器。CDConv的核心在于它的卷积核,该卷积核同时由当前正则位移和连续位移确定,由不同位移决定的核函数映射相对位置坐标,构建了一系列多尺度卷积核,来提取多尺度的蛋白质结构特征。对于表面编码器,由于以网格格式构建蛋白质表面需要相当长的时间,因此作者采用了dMaSIF中基于点云的蛋白质表面构建方法。该方法避开了表面静电等复杂的计算,只保留原始原子类型编码,可以实现基于光滑距离函数的在线表面构建。对于构建的蛋白质表面点云,作者利用基于KPConv的网络提取层次特征。除了卷积核之外,该网络与作者提出的结构编码器具有几乎相同的架构。与结构编码器一致,作者的表面编码器采用交替块和下采样来提取多尺度的特征。在每个尺度上,作者在这两个编码器的特征之间进行特征融合。与蛋白质结构和表面相比,蛋白质序列数据更容易获取。因此,许多基于序列的方法受益于对大型蛋白质序列数据集的预训练。这些预训练的基于序列的网络为蛋白质提供了有效的表征。因此,作者采用蛋白质语言模型ESM作为序列编码器。值得注意的是,Transformer中自注意力的二次复杂度要求截断输入的蛋白质序列。因此,由于潜在的部分遗漏,作者的序列编码器不能在氨基酸水平上与其他编码器完美对齐。为了解决这个问题,作者直接利用ESM提取的全局特征表示,而不是密集的氨基酸级表示。如图1所示,作者进行了两次融合处理。首先,在结构编码器和表面编码器之间进行多尺度双向融合;随后,作者将先前融合获得的特征与序列编码器提取的特征进行串联,进一步融合,生成特定任务的最终特征。具体来说,为了充分利用互补信息,作者提出了一种多尺度双向融合策略。与直接连接或级联不同,作者在每个尺度上进行双向融合。通过这种双向融合,结构和表面特征可以相互获得互补的信息,并演变成更加有效的特征。双向融合充分考虑了氨基酸和表面点之间的空间位置,选择那些接近的氨基酸和表面点进行融合。对于某些可能缺乏空间相邻表面点的内部氨基酸,作者用可学习特征代替相邻特征。最后经过融合层,所得特征既包含结构信息,也包含表面信息,将其放入平均池化层中,经过残差连接,得到特征表示。该特征与序列表示拼接融合,并用于后续的特定任务。作者将ProteinF3S与一些具有代表性的方法进行了比较,如表1所示,在基于不同酶反应类型的蛋白质分类任务上,ProteinF3S的准确度超越了现有方法。作者设计了消融实验来验证模型设计的有效性。在本节中,作者测试了仅使用序列、结构和表面编码器的性能,以及它们两两组合的性能。结果如表3所示。序列编码器和结构编码器的性能都非常好。前者得益于对大规模数据的预训练,而后者则实现了对几何结构的良好建模。表面编码器的性能相对有限。值得注意的是,两个编码器的任何组合都比以前产生更好的性能,这表明不同形式的蛋白质表示包含互补信息。更重要的是,ProteinF3S利用来自所有三种形式的互补信息,可以获得最佳性能。作者还进行了案例分析。如图2(a)所示,对于蛋白质表面,以蛋白质1a0i_a为例,它是蛋白酶中负责切割特定肽键的一部分。它的活性位点通常深埋在蛋白质中,依靠它的结构形成一个精确的催化袋。酶的功能与底物结合位点的内部结构密切相关,使得表面特征不足以捕捉其活性中心的关键属性。因此,表面编码器无法准确预测其酶反应类别。同样,许多酶表现出与内部和外部蛋白质结构相关的特征,这进一步解释了为什么表面编码器在这项任务中表现不佳。其次,对于蛋白质结构,以图2(b)中的蛋白106z_c为例。其高度复杂的褶皱,特别是与反应无关的螺旋或环等辅助结构,可能会给分类过程带来额外的噪音。这种复杂性阻碍了结构编码器捕捉酶反应的核心功能特征。相比之下,序列和表面形式提供了更简洁的信息。结果表明,表面编码器和序列编码器在预测中都取得了成功,而结构编码器则失败了。尽管这种结构形式似乎提供了最好的整体性能,但它仍然显示出某些缺点。最后,对于蛋白质序列,参考图4(c)中的5jis_d蛋白,它参与糖酵解途径。酶的活性位点依赖于三维折叠(红色和黄色分别代表α-螺旋和β-折叠)。由于序列本身不能直接反映蛋白质的折叠和空间结构,因此基于序列特征的预测在这种情况下是有限的。总的来说,每种形式的蛋白质表示都有自己的优点和缺点,融合多种蛋白质形式是合理和有效的。作者进一步对比结构和表面信息的不同融合策略,包括串联(CAT)、基于HOLOPROT的级联(Cascade)和单向多尺度融合(MSF,其中Struct2Surf表示结构到表面,Surf2Struct表示表面到结构),以及双向多尺度融合过程不使用残差连接(wo Res)。可以观察到,传统的串联和级联融合效果有限。这可能是因为表面编码器在本任务中的性能相对有限,影响了结构编码器。Surf2Struct在单向融合中优于Struct2Surf的性能也证实了这一点。然而,多尺度双向融合取得了积极的效果,证明了融合策略的重要性。此外,表3底部的两行也说明了融合过程中残差设计的必要性。残差的设计实现了选择性融合,使模型在合理的尺度和方向上传输互补信息。在这项研究中,作者提出了融合蛋白质序列、结构和表面之间存在的互补信息的框架ProteinF3S。此外,作者还提出了蛋白质结构和表面的多尺度融合策略,以进一步利用它们之间的互补信息。实验表明,融合不同形式的蛋白质确实可以提高性能,并且融合策略也起着至关重要的作用。未来,ProteinF3S有望扩展到更多的蛋白质任务,相关的融合策略也可以推广到更多类型的分子表征学习任务中。参考文献
Yuan et al. ProteinF3S: boosting enzyme function prediction by fusing protein sequence, structure, and surface. Briefings in Bioinformatics. 2025
内容中包含的图片若涉及版权问题,请及时与我们联系删除