


Predicting transcriptomes directly from genome sequences is a significant challenge in microbial genomics, particularly for the numerous sequenced microbes that remain unculturable or require complex experimental protocols like RNA-seq. The gap between genomic information and functional understanding leaves us without knowledge of the microbial adaptive processes, survival mechanisms, and gene regulation functions. This must be addressed to make better studies of microbial ecosystems, analysis of non-model organisms, and synthetic biology applications better.
Present techniques of transcriptome profiling are mainly experimental approaches such as RNA sequencing, which is time-consuming, expensive, and usually unsuitable for microorganisms with special growth requirements or those that survive under extreme environments. Computational models on UTRs or long DNA sequences are only partially useful since they can not be easily generalized to all taxonomic groups. Moreover, these methods fail to consider evolutionary constraints relevant to protein synthesis, making them even less useful in predicting transcriptomes of non-model and novel microbial species.
Researchers from the Beijing Institute of Technology and Harvard University propose TXpredict, a transformative framework for transcriptome prediction that utilizes annotated genome sequences. Leveraging a pre-trained protein language model (ESM2) extracts predictive features from protein embeddings while incorporating evolutionary principles. This innovation surmounts limitations on scalability, generalizability, and computational efficiency yet introduces new capabilities such as condition-specific gene expression predictions. Due to its capability to analyze the diversity of microbial taxa, including unculturable species, TXpredict is a significant advancement in microbial genomics.
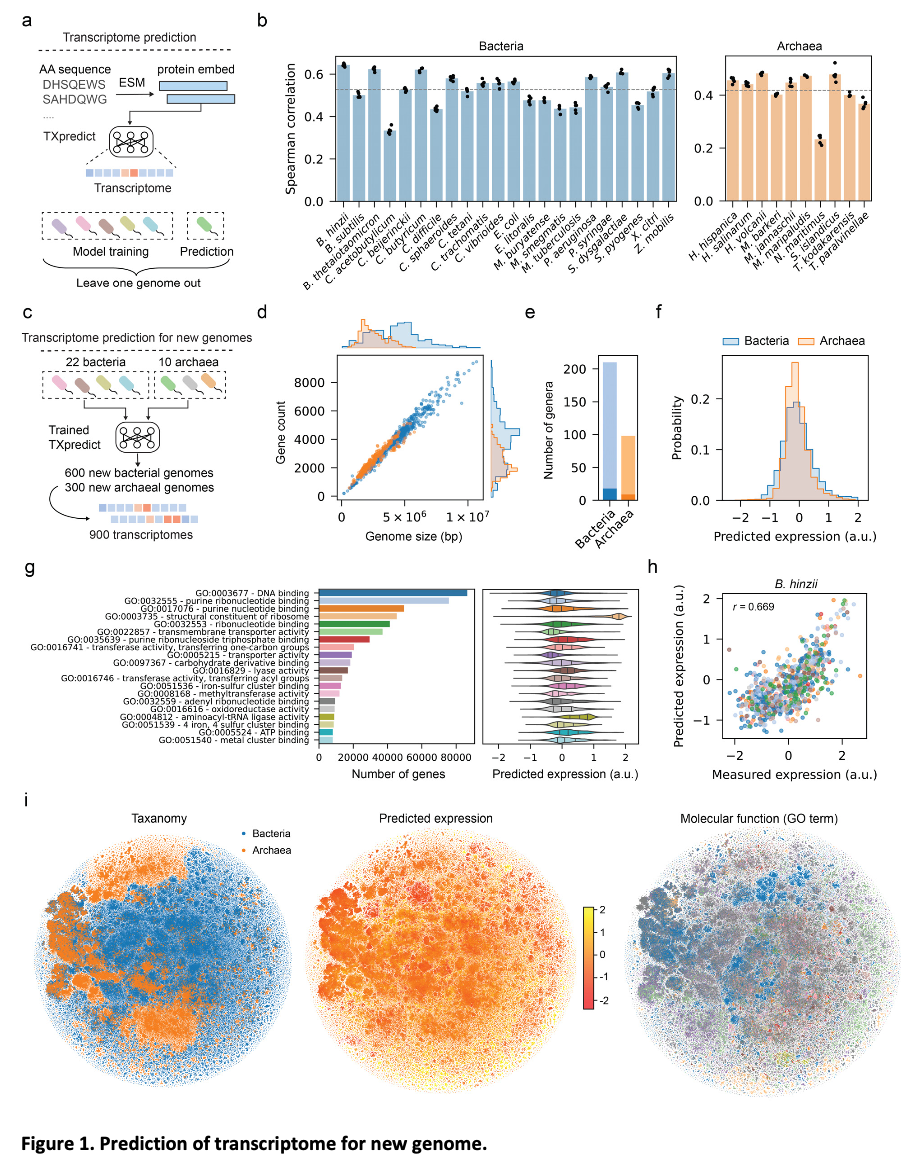
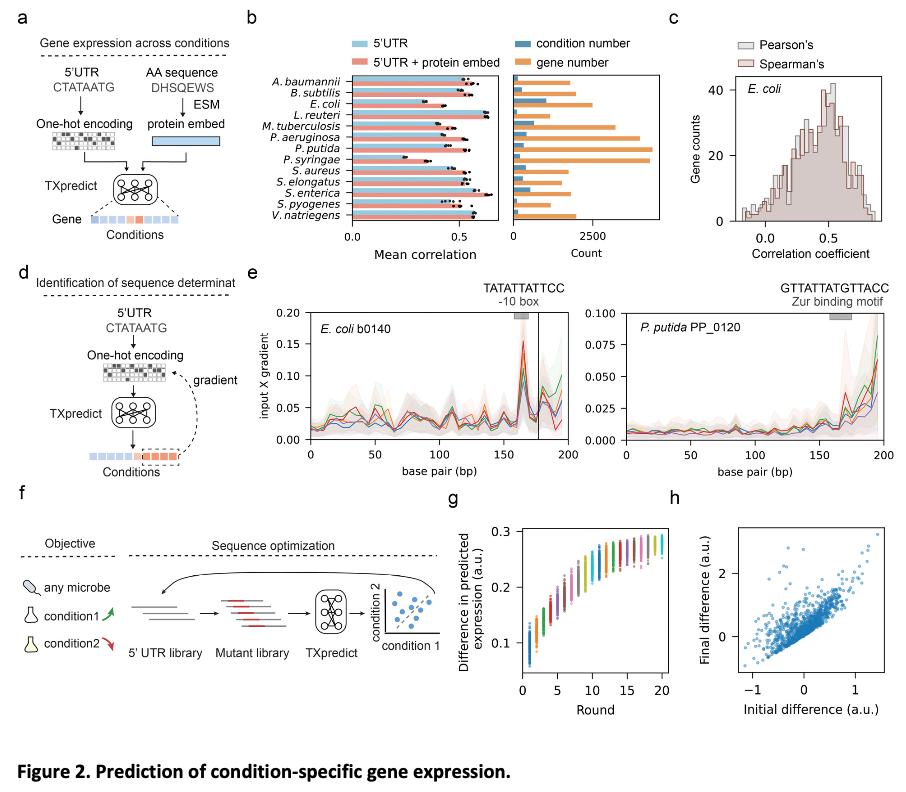
TXpredict is based on transcriptome data for 22 bacterial and 10 archaeal species, featuring 11.5 million gene expression measurements. The model uses a transformer encoder architecture with multi-head self-attention to capture complex sequence relationships. Inputs include protein embeddings from ESM2 and basic sequence statistics. Model training utilized leave-one-genome-out cross-validation for robust generalization. Condition-specific predictions were also enabled by incorporating 5′ UTR sequences. The framework is computationally efficient, completing transcriptome prediction for a microbial genome within 22 minutes on standard hardware.
TXpredict proved to be very accurate and scalable in the context of transcriptome prediction. It achieved a mean Spearman correlation coefficient of 0.53 for bacterial organisms and 0.42 for archaea and showed significant results for specific species such as B. hinzii (0.64), B. thetaiotaomicron (0.62), and C. beijerinckii (0.62). The predictions were extended to 900 additional genomes representing 276 genera and 3.11 million genes, which covered a large number of previously uncharacterized taxa. In the context of condition-specific transcriptomes, the model showed an average correlation of 0.52 over 4.6k experimental conditions, thereby capturing dynamic regulatory patterns. These results indicate that the framework is capable of giving precise predictions across a wide range of microbial species while keeping computational efficiency in check.
TXpredict addresses critical challenges in microbial genomics by bridging the gap between genome sequences and transcriptome predictions. This method, with the integration of protein embeddings, evolutionary constraints, and features specific to different conditions, presents a scalable, precise, and effective solution for various microbial taxa. This strategy not only yields valuable insights into gene regulation and adaptation but also possesses the potential to enhance synthetic biology and ecological research. Notwithstanding certain limitations, including dependence on pre-existing RNA-seq datasets and the exclusion of non-coding RNA components, TXpredict establishes a foundational framework for innovative applications in the field of microbial research.
Check out the Paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. Don’t Forget to join our 60k+ ML SubReddit.
 FREE UPCOMING AI WEBINAR (JAN 15, 2025): Boost LLM Accuracy with Synthetic Data and Evaluation Intelligence–Join this webinar to gain actionable insights into boosting LLM model performance and accuracy while safeguarding data privacy.
FREE UPCOMING AI WEBINAR (JAN 15, 2025): Boost LLM Accuracy with Synthetic Data and Evaluation Intelligence–Join this webinar to gain actionable insights into boosting LLM model performance and accuracy while safeguarding data privacy.
The post This AI paper from the Beijing Institute of Technology and Harvard Unveils TXpredict for Predicting Microbial Transcriptomes appeared first on MarkTechPost.
