


Protein language models (PLMs) have significantly advanced protein structure and function prediction by leveraging the vast diversity of naturally evolved protein sequences. However, their internal mechanisms still need to be better understood. Recent interpretability research offers tools to analyze the representations these models learn, which is essential for improving model design and uncovering biological insights. Understanding how PLMs process information can reveal spurious correlations, assess generalizability, and identify new biological principles. This analysis helps refine model biases and learning algorithms, ensuring reliability. Moreover, it sheds light on whether PLMs genuinely capture physical and chemical principles or merely memorize structural patterns.
PLMs, typically transformer-based, learn patterns and relationships in amino acid sequences through self-supervised training, treating proteins as a biological language. Prior studies have explored the internal representations of PLMs, using attention maps to uncover protein contacts and probing hidden states to predict structural properties. Research indicates that PLMs often capture coevolutionary patterns rather than fundamental protein physics. Sparse Autoencoders (SAEs) address the complexity of neuron activations by encoding them into sparse, interpretable features. This approach has improved understanding of neural circuits and functional components, offering insights into PLM behavior and enabling analysis of biologically relevant features.
Researchers from Stanford University developed a systematic framework using SAEs to uncover and analyze interpretable features in PLMs. Applying this method to the ESM-2 model identified up to 2,548 latent features per layer, many correlating with known biological concepts like binding sites, structural motifs, and functional domains. Their analysis revealed that PLMs often encode concepts in superposition and capture novel, unannotated features. This approach can enhance protein databases by filling annotation gaps and guiding sequence generation. They introduced InterPLM, a tool for exploring these features, and made their methods publicly available for further research.
Researchers employed SAEs to analyze latent features in PLMs using data from UniRef50 and Swiss-Prot. ESM-2 embeddings from transformer layers were processed, normalizing activations for consistent comparisons. SAEs were trained with 10,240 features using scalable parameters and validated against Swiss-Prot annotations with precision-recall metrics. Clustering methods like UMAP and HDBSCAN revealed interpretable structural patterns. For interpretability, features were linked to protein concepts using Claude-3.5 Sonnet for annotation. Sequential and structural analyses identified biologically significant patterns while steering experiments demonstrated how specific features could guide protein sequence generation. Methods and results are integrated into InterPLM for exploration.
SAEs trained on ESM-2 embeddings reveal interpretable features in PLMs. These features exhibit distinct activation patterns, identifying structural, protein-wide, or functional motifs. Unlike individual neurons, SAEs align better with Swiss-Prot concepts, showing stronger biological interpretability and covering more concepts. An interactive platform, InterPLM.ai, facilitates exploring these features’ activation modes, clustering similar features, and mapping them to known annotations. Features form clusters based on functional and structural roles, capturing specific patterns like kinase-binding sites or beta barrels. Additionally, automated descriptions generated by large language models like Claude enhance feature interpretability, broadening their biological relevance.
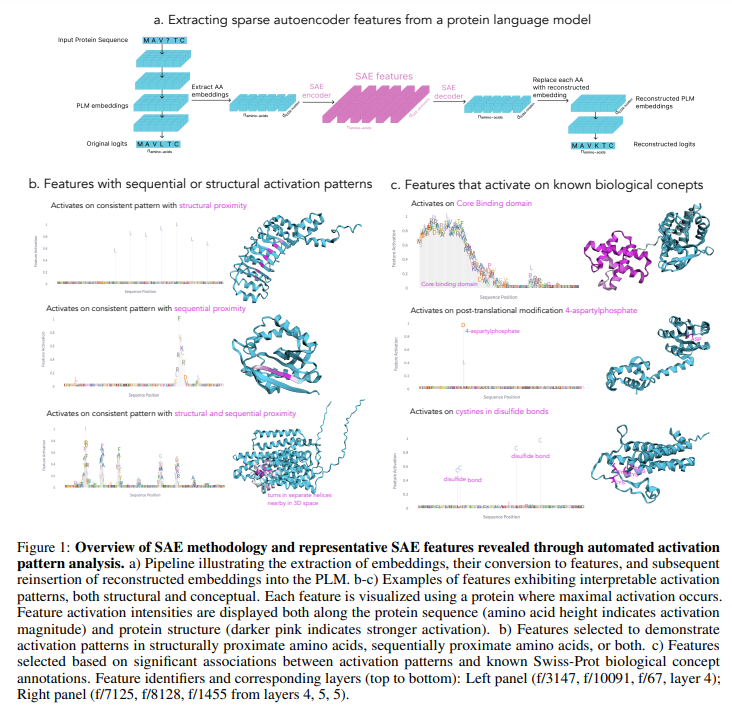
In conclusion, the study highlights the potential of SAEs to uncover interpretable features in PLMs, revealing meaningful biological patterns encoded in superposition. SAEs trained on PLM embeddings demonstrated superior interpretability compared to neurons, capturing domain-specific features tied to Swiss-Prot annotations. Beyond identifying annotated patterns, SAEs flagged missing database entries and enabled targeted control over sequence predictions. Applications range from model comparison and improvement to novel biological insights and protein engineering. Future work includes scaling to structural models, enhancing steering techniques, and exploring uncharacterized features, offering promising directions for advancing model interpretability and biological discovery.
Check out the paper. All credit for this research goes to the researchers of this project. Also, don’t forget to follow us on Twitter and join our Telegram Channel and LinkedIn Group. If you like our work, you will love our newsletter.. Don’t Forget to join our 55k+ ML SubReddit.
[FREE AI VIRTUAL CONFERENCE] SmallCon: Free Virtual GenAI Conference ft. Meta, Mistral, Salesforce, Harvey AI & more. Join us on Dec 11th for this free virtual event to learn what it takes to build big with small models from AI trailblazers like Meta, Mistral AI, Salesforce, Harvey AI, Upstage, Nubank, Nvidia, Hugging Face, and more.
The post Unveiling Interpretable Features in Protein Language Models through Sparse Autoencoders appeared first on MarkTechPost.
