
DRUGAI
今天为大家介绍的是来自MRC分子生物学实验室Sjors H. W. Scheres团队的一篇论文。如何处理持续弯曲的分子是冷冻电子显微镜(cryo-EM)图像中蛋白质单颗粒分析中最重要的挑战之一。在本文中,作者介绍了DynaMight,这是一款通过学习共识结构的高斯伪原子模型的三维变形来估计cryo-EM数据集中连续构象空间的软件工具。随后,通过对学习到的变形进行反转,以获得改进的共识结构重建。作者展示了DynaMight在多个实验性cryo-EM数据集中的表现。作者还展示了如何通过在cryo-EM数据的两个半集中独立训练两个变分自编码器来获得变形的误差估计,以及如何通过使用原子模型对三维变形进行正则化可能会因模型偏差导致重要的伪影。

通过cryo-EM图像的单颗粒分析确定生物大分子结构,本质上是一种单分子成像技术。cryo-EM数据集中多个单独复合物的图像包含了样本在快速冷冻时的全部分子动力学信息。然而,为限制辐射损伤而采用的低剂量成像条件会导致较高的实验噪声,需要对多个图像进行平均处理,以提取大分子三维结构的详细信息。然而,平均处理不同结构的投影图像会导致三维重建的模糊,通常使用图像分类算法将数据集分成用户定义的结构同质子集。
尽管这些算法在处理离散构象方面有效,但面对样本中的连续分子运动时却存在挑战,连续分子运动往往被视为干扰,而非蛋白质动力学的重要信息。早期的一种尝试是使用流形嵌入描述cryo-EM数据集中的连续分子运动,但该方法的应用范围有限。另一种较广泛使用的方法是多体微调,将复合物分成独立运动的刚体,通过部分信号减法来改善图像的对齐和重建效果。近年来,深度卷积神经网络的变分自编码器(VAE)被用于将投影图像映射到连续多维潜在空间,从而能够在潜在空间中重建三维结构,并生成描述蛋白质运动的动画。
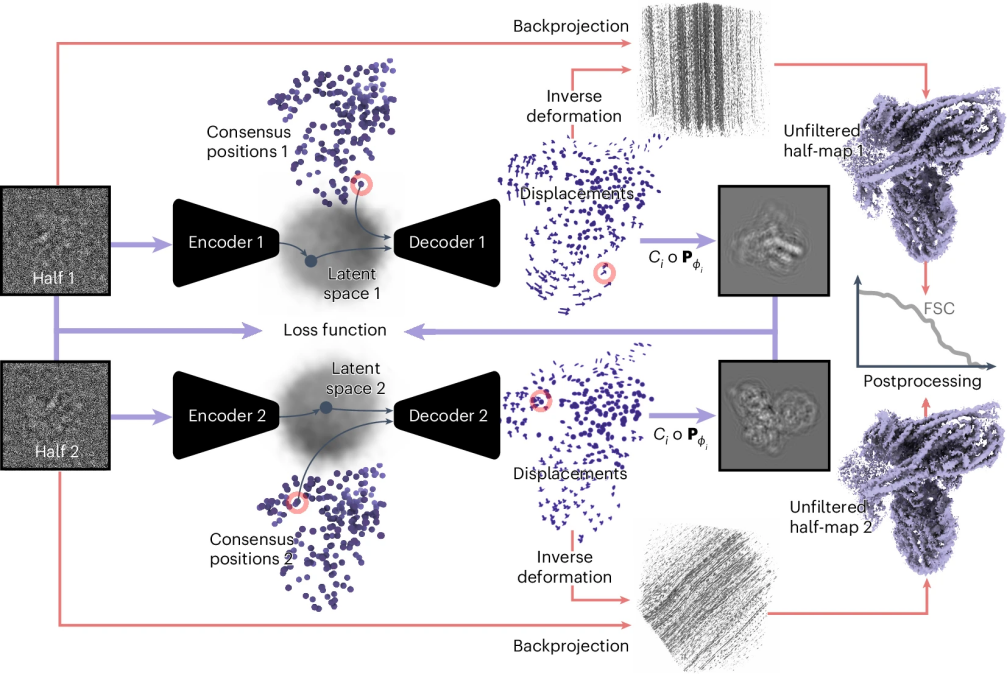
图 1
如图1所示,本文提出了DynaMight方法,它利用高斯伪原子建模cryo-EM密度,使用VAE估计数据集中的构象变化。作者引入了一种解码器架构,直接输出高斯伪原子的三维变形场,从而可以在变形场中直接应用先验知识,最终通过改进的反投影算法生成共识结构的改进密度图。
构象变异描述
作者用以下前向模型来描述第Nd个粒子图像yi:
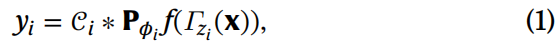
作者假设所有粒子图像都是单一共识结构的构象变化,该结构由Ng3D高斯基函数描述,公式(1)中的zi是第i个图像的构象编码。作者将个别粒子的变形描述为相对于共识坐标x的偏差:Γ(x)=x−δ(x),因此:
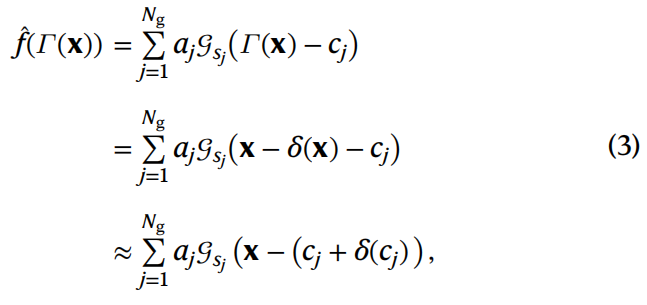
最后的近似假设变形场在局部是恒定的,并且cj周围的密度以相似方式移动。这使得能够将变形描述为高斯中心的位移,这是一种计算上可行的表示方法。此外,所有高斯伪原子的宽度sj和幅度aj在整个数据集中保持不变。这意味着DynaMight的设计只能模拟质量守恒的异质性,不能处理非化学计量的混合物。
因此,在运行DynaMight之前,应通过其他方法从数据集中去除组成异质性。
构象变异评估
如图1所示,为了学习这些变形,作者使用了一个由两个神经网络组成的VAE(变分自编码器),分别是一个编码器E,它为每个粒子图像预测一个l维的潜在表示zi,以及一个解码器D,它预测模型中所有高斯伪原子的位移。
编码器是一个具有三层线性层和ReLU激活函数的全连接神经网络。输入是一个(实空间)实验图像yi,输出是两个向量(μi ,σi)∈RNl×RNl,描述了用于生成样本zi的均值和标准差,这个样本将作为解码器的输入。解码器D(zi ,cj)近似每个zi对应的cj+δj项。作者为整个Ng个位置定义了解码器:
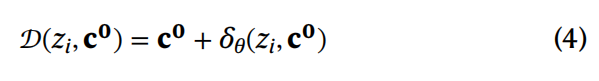
这些输出位置用于生成粒子姿态下变形模型的投影图像pi,并在神经网络训练期间通过最小化投影图像与实验图像之间的差异 ||pi−yi|| 2Σ 来优化网络。一旦训练完成,针对整个数据集的潜在嵌入,就可以获得定义在整个三维空间上的一系列变形场D(zi,x) ≈ Γzi(x)。
正则化与模型偏差
由于实验噪声较高,cryo-EM重建是一个病态问题。即使对于标准的结构同质的微调,每个图像也有许多可能的旋转和平移分配。在估计构象变异时,虽然姿态已知,但许多变形的密度图同样可以解释每个实验图像。因此,在这两种情况下,正则化对于稳健的重建至关重要。
为了探索变形场的直接正则化,作者测试了两种方法。第一种方法是在运行DynaMight之前,使用构建在共识图中的原子模型的先验信息。它生成原子位置的粗粒化高斯表示,然后根据原子模型中的键存在情况最小化这些高斯之间的距离变化:
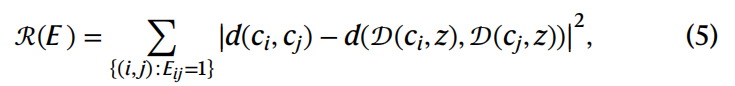
其中,当伪原子ci和cj之间存在键时,Eij=1,而d表示欧氏距离。使用这种正则化方案的变形会使高斯保持接近于原始原子模型的粗粒化表示。
第二种正则化方法使用较少的先验信息,不需要原子模型。相反,随机放置高斯以填充共识图中的密度,并且方程(5)中的连接E是在所有高斯对之间建立的,其距离为所有高斯及其两个最近邻之间平均距离的1.5倍。此正则化方法强制变形的整体平滑性。此外,还有额外的惩罚项,防止高斯彼此过于接近或离其他高斯太远,以确保高斯分布的物理合理性。
改进后的3D重建
作者提出了一种算法,利用估计的变形场Γ来获得包含所有实验图像信息的改进共识结构重建。要将单个粒子图像映射回假设的共识状态,需要估计逆变形,这是一个挑战。对于位移后的高斯函数,逆变形由负位移向量给出,即Γ−1(Γ(ci))=ci,但在改进的重建中,逆变形场需要在所有笛卡尔网格点上进行推断。为此,作者训练了一个神经网络作为回归函数,以估计在给定采样点Γ(ci)上与变形场一致的变形场,并可以在任意位置进行评估。该网络由一个包含六层的多层感知器和一个与共识模型c0的原始坐标的单一残差连接组成。与前向变形模型类似,网络以潜在编码zi和变形后的位置Γ(ci)作为输入,目标是输出原始位置ci。除了在采样点上进行前向场的逆转外,作者通过在损失函数中添加正则化项来强制逆场的平滑性。
正则化可能导致模型偏差
作者首先在一个针对酵母Saccharomyces cerevisiae的前催化B复合体剪接体数据集(EMPIAR-10180)上,分析了不同的变形正则化选项。
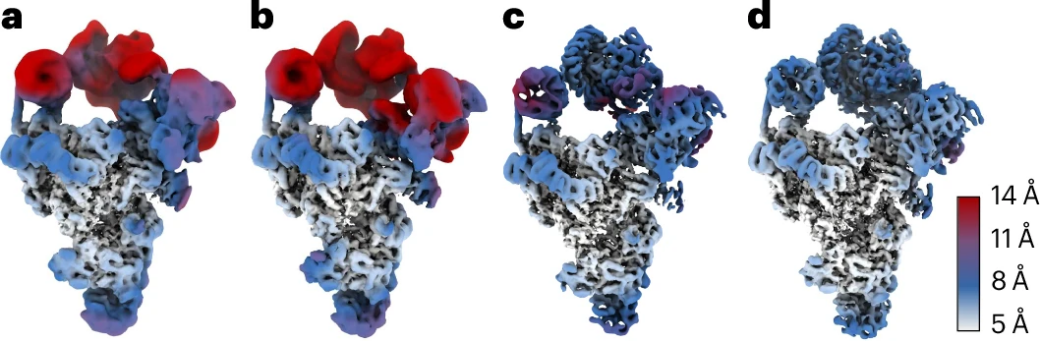
图 2
在没有对变形进行任何正则化的情况下,估计的变形场显示出相邻高斯函数的方向快速变化,且变形反投影得到的重建在局部分辨率方面并没有比原始共识重建有所改善(图2a, b)。图2c通过使用强制变形平滑的正则化方案但不使用原子模型,得到了局部分辨率更好的共识重建。图2d中使用强制原子模型中键合原子间距离的正则化方案,获得了最高的局部分辨率。这表明将原子模型的先验知识引入VAE是有益的。
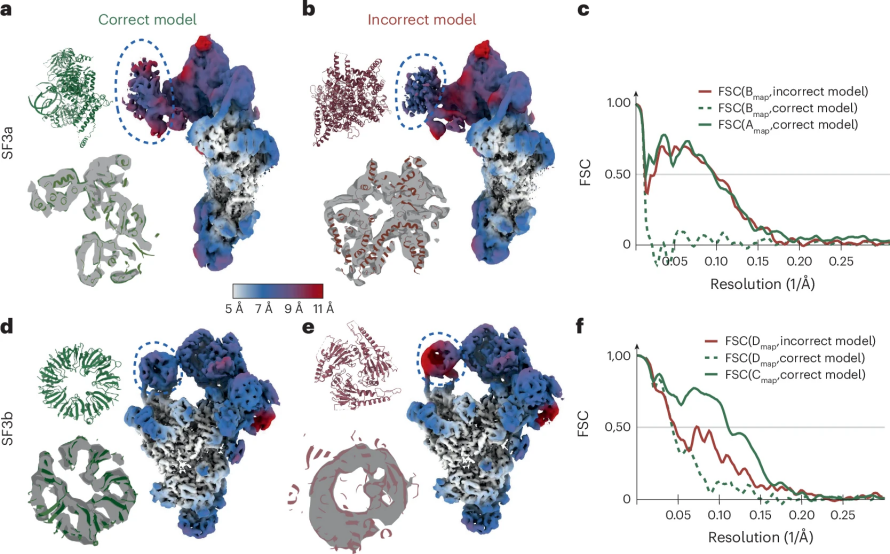
图 3
在第一个对照实验中,作者用一个不同但大小相似的蛋白质域(PDB 7YUY)替换了U2 3′域/SF3a域的原子模型。如图3a, b所示,尽管使用不正确的原子模型来估计变形场,在局部分辨率方面与使用正确模型相比有类似的改善,但图3c中变形反投影重建的密度更像是不正确的模型,而不是正确的模型。
在第二个对照实验中,作者用PDB 1G88替换了SF3b域的原子模型。共识图中SF3b域的密度比SF3a区域的密度更强,表明该区域在剪接体中不太灵活。在这种情况下,使用不正确的原子模型在SF3b区域生成的地图的局部分辨率低于使用正确模型生成的地图(图3d, e)。但即便如此,图3f中变形反投影重建的密度仍然更接近不正确的模型,而非正确的模型。
这些结果表明,变形场的估计可能导致模型偏差,以至于重建的密度可能会再现不正确的原子模型的特征。在具有较高结构异质性的地图区域,模型偏差对变形反投影重建的影响更大。
DynaMight改善了内着丝粒复合体的图像质量
接下来,作者在两个酵母内着丝粒的cryo-EM数据集上展示了DynaMight的实用性。
第一个数据集EMPIAR-(11910)包含100,311个结合在CENP-A核小体上的单体型结构性着丝粒相关网络复合物(CCAN–CENP-A)粒子。
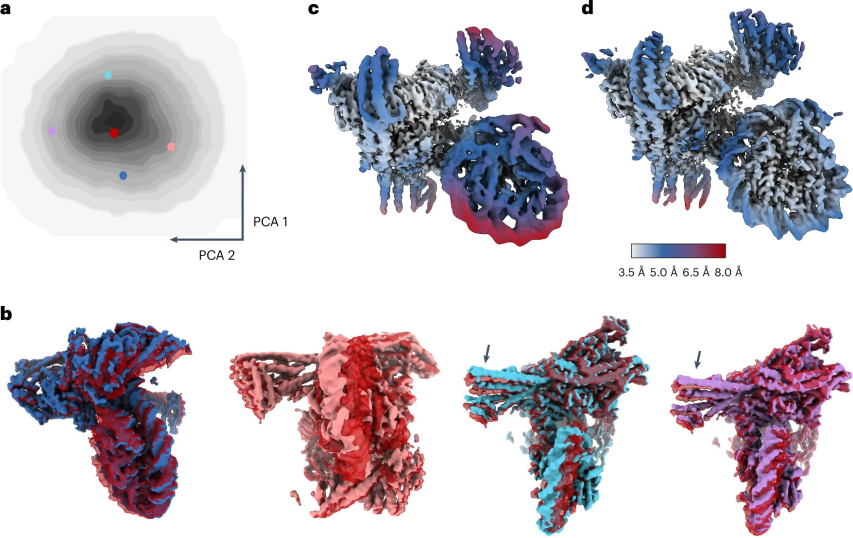
图 4
如图4a所示,估计的3D变形均匀分布在潜在空间中没有特定的聚集构象状态,这表明数据集中的运动主要是连续的。图4b的运动分析显示,相对于复合物的其他部分,核小体在不同方向上旋转,这些旋转与Nkp1、Nkp2、CENP-Q和CENP-U亚基的上下弯曲共存。与标准RELION微调的共识图相比,变形反投影的重建改善了局部分辨率,蛋白质和DNA的特征都有明显的改进(图4c, d)。
第二个数据集EMPIAR-(11890)包含108,672个组装在CENP-A核小体上的完整酵母内着丝粒复合物粒子。
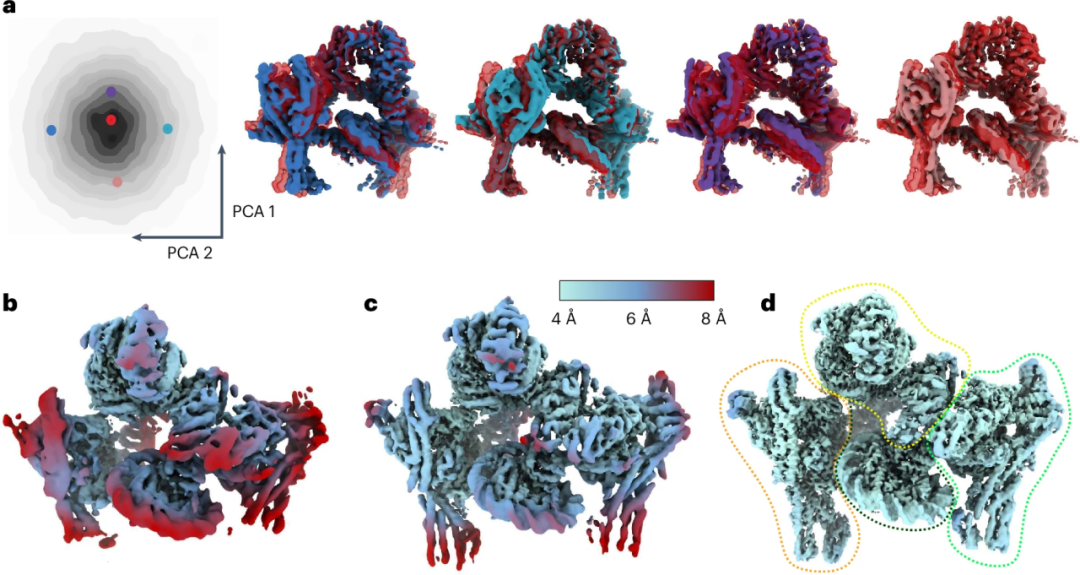
图 5
同样,如图5a所示,潜在空间中的变形连续分布表明结构具有连续的灵活性。变形分析揭示了复合物不同区域之间的大幅相对运动。变形反投影导致的地图相比共识微调映射,在局部分辨率和蛋白质与DNA特征上都有所改善(图5b, c)。
讨论
如何处理连续构象异质性仍是cryo-EM单颗粒分析中的一个快速发展的主题。本文提出的DynaMight方法使用独立训练的两个VAE来估计高斯模型的位移,并通过加权反投影算法修正估计的变形。然而,DynaMight无法处理组成异质性,建议在应用前通过离散分类方法去除异质性。尽管DynaMight在提高具有高度灵活性的分子复合物的cryo-EM图像方面表现出色,但仍存在进一步改进的空间,特别是在处理连续结构异质性方面。未来研究应关注如何利用先验知识避免模型偏差,同时提高变形估计的验证方法。
编译 | 于洲
审稿 | 曾全晨
参考资料
Schwab J, Kimanius D, Burt A, et al. DynaMight: estimating molecular motions with improved reconstruction from cryo-EM images[J]. Nature Methods, 2024: 1-8.
