
DRUGAI
今天为大家介绍的是来自邓岳/戴琼海团队的一篇论文。近年来,空间组学技术的进步已经扩展了被分析的分子类别范围,不再局限于转录组学。然而,这些技术中的许多由于空间分辨率有限,限制了我们深入表征复杂组织结构的能力。现有的计算方法主要集中于提高转录组学数据的分辨率,但缺乏应对新兴空间组学技术的适应性。在此,作者介绍了一种名为soScope的统一生成框架,旨在提升从各种空间技术中获得的分子谱的质量和空间分辨率。soScope通过分布先验进行特定组学的建模,整合了来自组学、空间关系和图像的多模态组织信息,并联合推断出增强分辨率的组学谱。在Visium、Xenium、spatial-CUT&Tag和slide-DNA/RNA-seq等多种空间组学平台上的全面评估中,soScope提高了在识别具有生物学意义的肠道和肾脏结构、揭示在原始分辨率下无法解析的胚胎心脏结构以及校正因测序和样本处理引起的样本和技术偏差方面的性能。此外,soScope还扩展应用于spatial-CITE-seq和spatial ATAC-RNA-seq等空间多组学技术,通过利用跨组学的参考实现同时增强多组学数据。

组织是由具有不同分子状态和空间组织的细胞构成的。要深入剖析组织结构,需要在保持其空间背景的同时,对多种分子谱进行深度表征。近年来,空间组学技术的重大进展使得我们能够对各种分子类别进行空间谱分析,包括转录物、蛋白质、表观遗传标记以及基因组变异。通过揭示多种分子谱的空间特征,空间组学在癌症、阿尔茨海默病和胚胎发育等多个生物学领域提供了宝贵的见解。
尽管现有的空间组学方法已取得初步成功,但仍存在两个限制其全面潜力的挑战。首先,在测序之前,组织通常处于冷冻或福尔马林固定和石蜡包埋(FFPE)的状态。冷冻过程和福尔马林诱导的脱氨作用可能会影响分子的状态,导致测序准确性降低。其次,大多数空间技术使用空间条形码标记目标分子的位点,并在组织点分辨率上进行组学分析。由于每个点通常包含数十个细胞,多细胞平均测序读数减少了可解析的异质性,导致组织结构的空间分辨率受限。
计算技术有望减少数据变异性并提高空间组学数据的空间分辨率,从而促进更精确的组织结构表征。然而,目前的方法主要针对单一组织模式的组学谱进行增强,如空间位置(例如 BayesSpace)或图像(例如XFuse和iStar),这忽略了大多数空间平台中丰富的多模态组织信息。此外,这些方法主要为转录组数据量身定制,其统计假设无法扩展到其他空间组学类别。
soScope模型架构
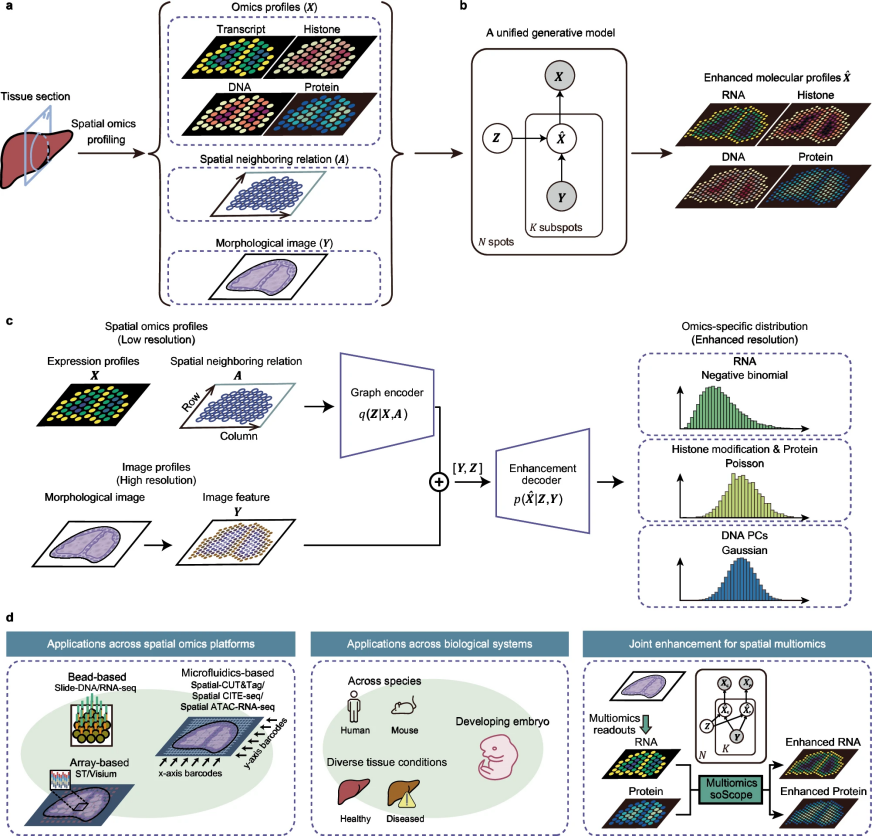
图 1
在此,作者介绍了一种称为空间组学范围(soScope)的完全生成框架,该框架对来自不同空间组学技术的点级谱生成过程进行建模,旨在提高其空间分辨率和数据质量(图1a)。为实现这一目标,soScope将每个点视为增强空间分辨率下的“亚点”集合,其组学谱与空间位置和形态模式相关联(图1b)。然后,soScope 通过多模态深度学习框架整合点组学谱、空间关系和高分辨率形态图像,共同推断亚点分辨率下的组学谱。通过选择特定组学的分布,soScope能够准确建模并减少不同空间组学数据的变异(图1c)。
具体地说,soScope是一个统一的生成模型,能够整合来自不同空间组学平台的分子谱(X)、空间邻近关系(A)和形态图像特征(Y),其目标是将每个点划分为多个亚点(X^)。该模型包括三个步骤:
点级别表示学习:在原始分辨率下,分子谱及其空间坐标通过图编码器q(Z|X, A)被嵌入到潜在表示(Z)中;
亚点级别图像特征学习:对应亚点区域的图像片段从高分辨率的组织图像中分割出来,并转换为亚点的形态特征;
亚点级别组学谱推断:点表示和亚点图像特征在增强解码器p(X^|Y, Z)中结合,共同推断出受组学特定分布和形态相似性约束的亚点级别谱。
图编码器为点级组学谱提供了高质量的嵌入,同时保留了空间邻近信息。图像特征是从精确的亚点区域中提取的,并提供了增强分辨率的信息,以指导相应组学谱的推断。增强解码器通过反映其统计变异的概率分布,准确地对多种亚点组学数据进行建模。通过优化其证据下界(ELBO),soScope高效生成了具有降低数据噪声和增强空间分辨率的组学谱,从而对组织结构进行更精细的表征。
soScope在空间转录组学增强方法中表现优于其他方法
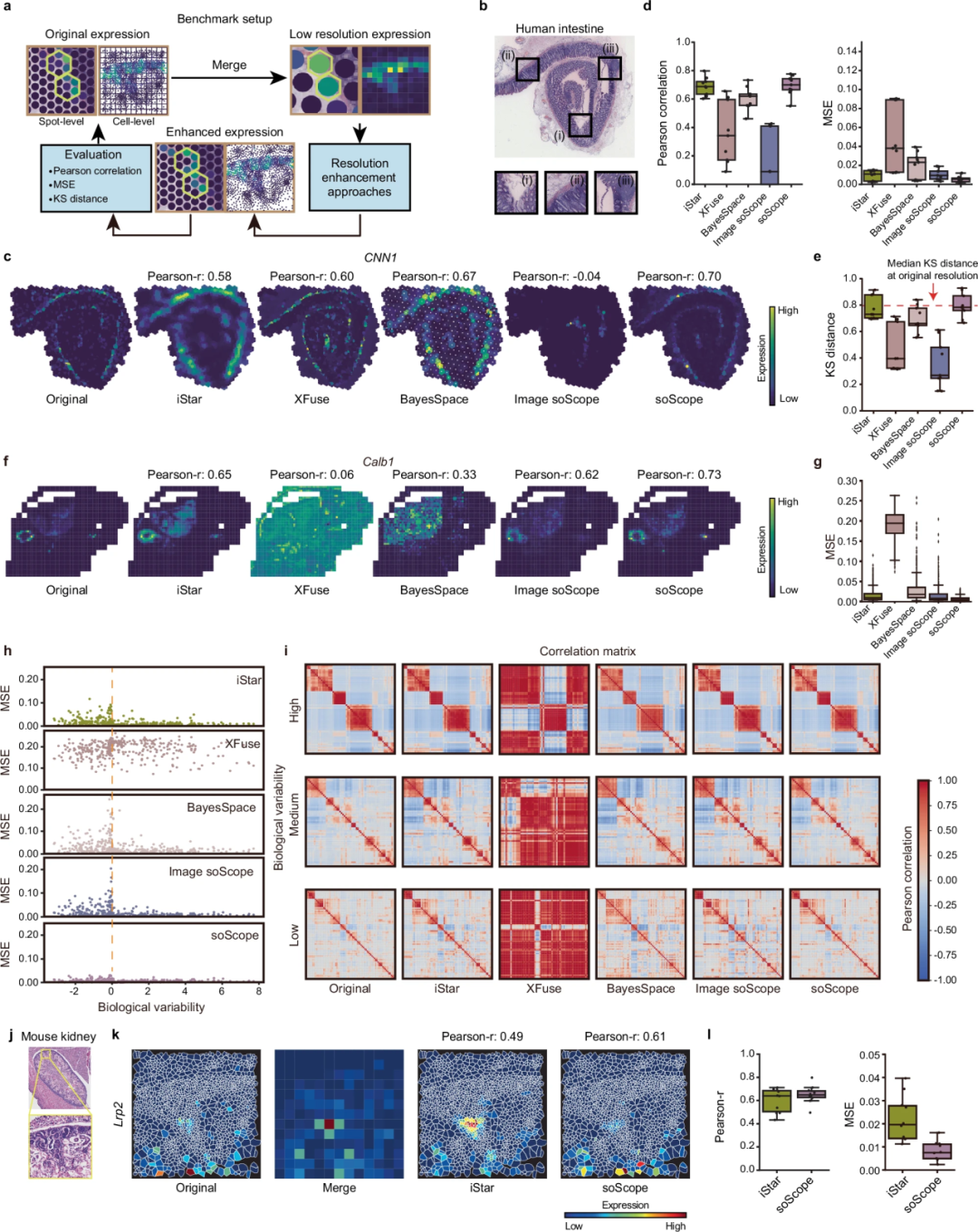
图 2
现有的计算方法主要集中于提高空间组学的分辨率,特别是针对转录组学。因此,作者首先在由不同平台生成的空间转录组学(ST)数据集上评估了soScope。作者将它与三种已有的方法进行了比较:利用图像信息增强转录谱的iStar和XFuse,以及使用空间坐标推断高分辨率表达数据的BayesSpace。此外,作者还将一个只使用图像输入的soScope变体作为参考,称为image soScope。由于缺乏高分辨率的真实数据进行评估,作者通过合并相邻点的转录数据来模拟“低分辨率”表达谱。如图2a所示,作者通过将恢复的表达与原始谱进行比较来评估分辨率增强方法的性能。
首先,作者考虑一个由Visium平台生成的人类肠道数据集,该数据集包含2,649个测序点,并配有高分辨率的苏木精-伊红(H&E)染色图像。从增强分析来看,仅利用图像信息的iStar在免疫区域表现良好,但它未能恢复上皮和肌层区域的精细结构;另一种基于图像的方法XFuse在肌层区域表现良好,但由于区域之间强烈的形态相似性而被误导,未能忠实保留上皮和免疫区域的组织结构;仅依赖空间信息的BayesSpace并未实质性地细化增强谱中的区域边界;由于缺乏转录组信息,image soScope未能恢复大多数模式。相比之下,通过使用Pearson相关系数和均方误差(MSE)对增强表达进行定量评估,soScope表现出最高的一致性和最低的重建误差(图2d)。接下来,作者使用 Kolmogorov-Smirnov(KS)距离评估每个标记基因在其对应组织区域内外的表达分布差异。soScope和iStar在大多数测试基因上显示了最显著的分离(图2e),并实现了与原始谱相似的区分能力(红色虚线)。
其次,作者评估了从Xenium平台获得的小鼠头部数据集。从增强结果来看(图2f, g),作者发现iStar和soScope都在保留高分辨率表达谱方面表现良好。通过根据基因的变异性(图2h)或丰度进行检查,作者发现soScope在不同变异性情况下表现更为稳定。此外,作者还调查了分辨率增强是否会影响基因相关性。如图2i所示,除了XFuse,不论生物变异性差异如何,大多数方法在分辨率增强后仍能保持基因间的相关模式。
第三,作者评估了soScope在单细胞分辨率增强中的能力。作者使用了一个由Xenium平台生成的小鼠肾脏数据集(见图2j),该数据集覆盖1,538个细胞。作者将组织划分为小的方形区域,并聚合每个区域内细胞的基因表达,以恢复其细胞表达。结果表明,iStar倾向于高估高细胞密度和高表达区域的表达水平(见图2k)。相反,soScope表现出与真实值更为一致的表达模式(见图2l)。
这些发现表明,soScope 在跨越各种技术平台、分辨率需求和基因统计特征的转录数据中,能够增强空间分辨率,促进组织结构的详细表征。
soScope减少测序噪声并揭示小鼠胚胎的详细结构

图 3
近年来,空间组学技术的进步扩大了在组织中分析分子信息的能力,超越了传统的转录组学。soScope可以灵活地扩展,以结合生成过程和特定组学的分布来处理不同的组学类型。为了展示这种多功能性,作者将soScope应用于由spatial-CUT&Tag生成的空间染色质可及性数据集。该数据是在胚胎发育第11天的小鼠胚胎上收集的,包括1974个按照50×50阵列排列的测序点以及H&E染色图像。作者从四个主要器官区域(肝脏、心脏、前脑和脊髓)中选择了60个最具变异性的峰进行分辨率增强分析(图3a)。
由于染色质可及性数据没有现成的增强方法,作者改编了六种通用的机器学习模型进行增强比较:
基于空间的方法:
使用空间最近邻(Spatial linear)或基于空间坐标的高斯过程映射(Spatial GP)来估计增强分辨率的谱;
基于图像的方法:使用线性回归器(Image linear)、高斯过程模型(Image GP)或多层感知器模型(Image MLP)在亚点分辨率下从图像特征中预测谱;
多模态联合方法:使用多层感知器模型(Joint MLP)通过图像和空间位置的组合特征来增强组学。
作者首先通过模拟研究评估了性能。具体而言,作者将每2 × 2个相邻点合并为一个“低分辨率”点,以生成“低分辨率”的峰值数据。然后,作者尝试使用上述分辨率增强方法恢复原始谱。soScope在保持原始分辨率的峰值计数方面实现了最高的相关性和最低的重建误差(图3b)。
接下来,作者将组织分辨率从原始数据直接提高了4倍(从1,974个点到7,896个增强亚点)。当对数据应用分辨率增强时,仅考虑空间位置的方法(Spatial linear和Spatial GP)基于原始的噪声数据插值了不准确的峰值计数;同时,基于图像数据预测的方法(Image linear、Image GP和Image MLP)受到随机噪声的困扰未能捕捉到目标区域中的表达模式,特别是在心脏和脊髓中(图3c)。图像和位置的直接组合方法(Joint MLP)确实提供了一定的改进。相比之下,soScope结合了相邻点之间的空间关系和图像参考正确识别了表达区域,同时有效地抑制了区域外的表达。
组织谱的增强使得可以在更精细的分辨率下研究组织结构。从放大的H&E图像中可以看出(图3d左侧, 用黄色虚线分隔),心脏区域可以进一步分为两个子区域:小梁心室心肌和致密心室心肌。当可视化该区域中对应于两个已知标记基因的谱峰时(Fhl2对应小梁心室心肌,Ldha对应致密心室心肌),由于低分辨率,它们的强度未能形成可识别的模式(图3d右侧)。在分辨率增强后,作者可视化了通过比较方法预测的空间峰值活性(图3e)。正如预期的那样,基于空间的方法未能改善结构表征。在其他方法中,soScope增强的峰值谱更好地反映了心脏的双层结构,Fhl2基因在小梁心室心肌中显示出高活性,而Ldha在致密心室心肌中表现出高活性。作者利用了一个来自人类胚胎的心脏ST数据集(图3f左侧),并交叉检查了这两个基因的空间表达模式(图3f右侧)。FHL2在小梁心室心肌中表现出高表达,而LDHA主要富集在致密心室心肌层中表现出高表达,这与soScope增强的spatial-CUT&Tag数据中获得的基因活性分数一致。定量分析进一步证实了基因表达与染色质可及性活动之间分布模式的相似性(图3g)。
soScope在从模拟的极低分辨率数据中恢复组织结构
由于不同的空间组学技术具有不同的空间分辨率,作者设计了一项多尺度测试,以评估和校准不同方法在增强不同原始分辨率的组学数据时的性能。
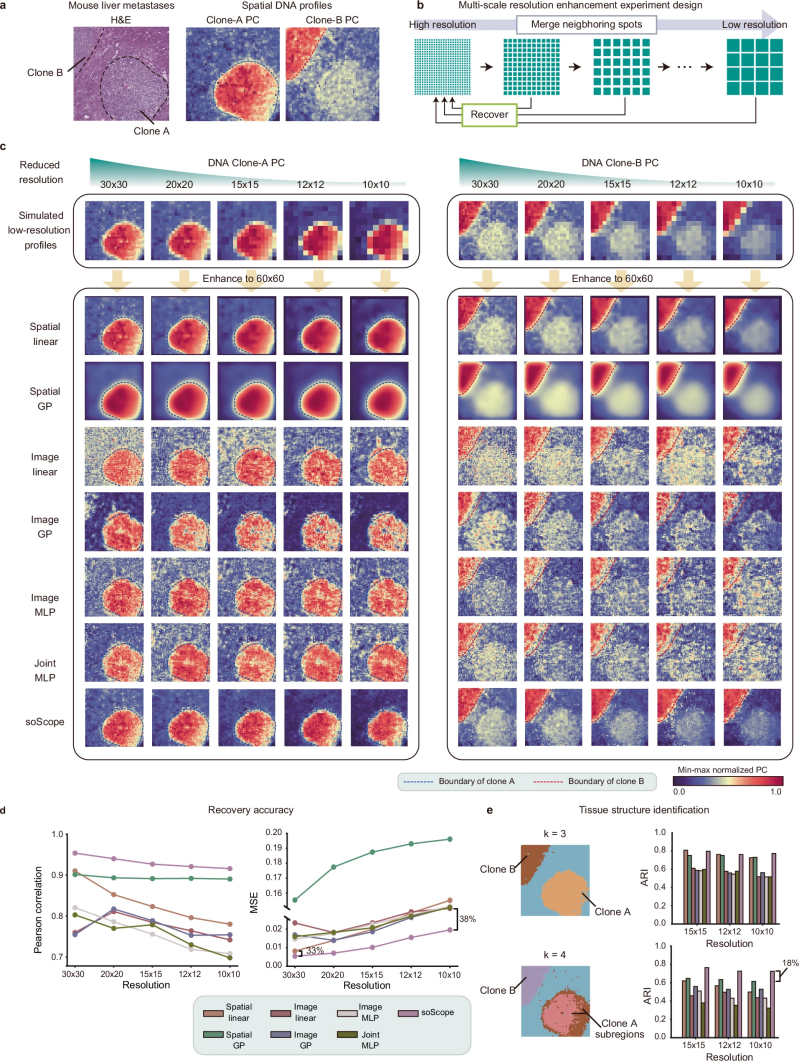
图 4
为了进行多尺度测试,作者使用了一个由slide-seq获得的小鼠肝肿瘤数据集,并实现了接近单细胞分辨率的空间测序。slide-seq原始数据的高分辨率允许在不同尺度上生成低分辨率数据,并测试从中恢复原始谱的能力。肝肿瘤组织包括两个转移克隆区域,并使用slide-DNA-seq和slide-RNA-seq进行了分析,分别包括24,679个DNA heads和31,286个RNA heads(图4a)。为了进行测试,作者将整个组织划分为60 × 60分辨率的小方块区域,并逐步将不同大小的相邻区域合并为一个低分辨率点。使用所有测试的方法,作者的目标是从降低分辨率的数据中恢复原始分辨率的谱(图4b)。
作者在soScope模型中使用高斯分布来建模DNA主成分。在原始数据中,DNA主成分1在克隆A区域(Clone-A PC)中高度富集,而主成分2在克隆B区域(Clone-B PC)中富集。随着分辨率的降低,DNA区域模式显著退化(图4c第一行)。在分辨率增强之后,作者发现仅考虑点之间空间关系的方法(Spatial linear和Spatial GP)能够捕捉到与两个癌症克隆相对应的正确区域;然而,这些方法在从极低分辨率(10 × 10)的数据中恢复时,往往会过度平滑表达模式。相比之下,基于图像或联合预测的方法(Image linear、Image GP、Image MLP和Joint MLP)更好地保留了局部表达模式。然而,这些方法被H&E数据误导,重建的高分辨率DNA主成分更接近于H&E图像定义的克隆区域,而不是原始DNA数据(图4c左侧;与虚线区域轮廓相比)。即使是从极低分辨率的数据中,soScope也成功地保留了局部表达模式,同时准确地重建了原始克隆区域。这凸显了soScope在使用生成框架整合空间和跨模态信息以实现稳健分辨率增强方面的重要性。
从定量评估中(图4d),大多数方法在分辨率降低时精度如预期般下降。soScope在不同分辨率下表现出稳定的性能,精度下降相对较小。为了进一步验证重建的DNA特征是否能够反映组织结构,作者对增强的谱进行了k-means聚类分析。当作者将聚类数设置为3时,所有方法都成功识别出了先前定义的三个区域(图4e上方)。当将聚类数增加到4时,揭示了克隆A中一个由主成分1高度表达特征的子区域(图4e下方)。在各种方法中,只有soScope和Spatial GP重建的DNA主成分在所有降低分辨率的情况下始终能够一致地捕捉到这一子结构。这凸显了soScope在准确揭示各种组学类型和空间分辨率下组织潜在空间结构方面的稳定表现。
Multi-soScope通过跨组学参考校正技术偏差
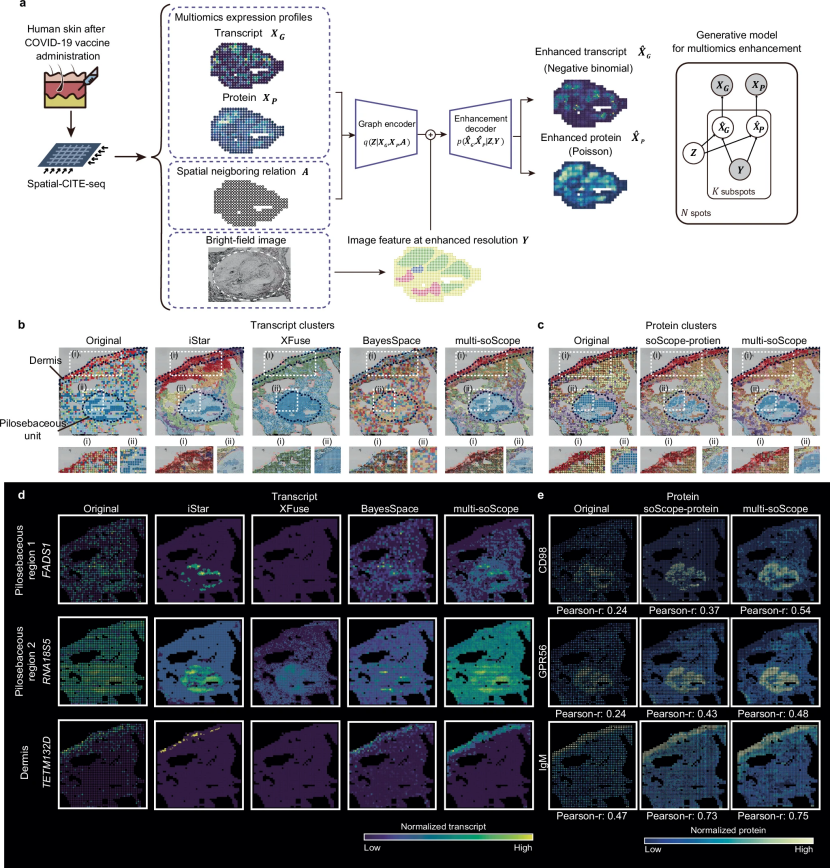
图 5
近年来,空间多组学技术的进步扩大了对组织进行空间分析的能力,超越了单一组学类型。作者通过以下修改使soScope能够与空间多组学数据一起工作(称为multi-soScope, 图5a):
构建图编码器时,空间图基于转录物和蛋白质的相似性,提供了空间异质性的综合度量;
转录物和蛋白质模态的特征同时输入编码器,以生成联合的潜在表示,从而实现两种空间组学谱信息的整合;
解码器同时输出增强的多组学谱的分布参数,允许对多组学变异进行同时建模。具体而言,对于spatial-CITE-seq的应用,使用负二项分布和泊松分布分别对增强的转录物和蛋白质谱进行建模;对于spatial ATAC-RNA-seq,使用负二项分布和高斯分布对增强的转录物谱和归一化的ATAC峰进行建模。
作者将soScope应用于一个在COVID-19 mRNA疫苗注射后从人类皮肤组织中收集的spatial-CITE-seq数据集。如图5b所示,从原始数据中,由转录物聚类识别的亚群显示出高水平的噪声。使用现有的转录组学方法进行分辨率增强后,增强的转录物谱受到原始数据质量的影响:iStar和XFuse都在真皮层区域周围简化了组织结构;BayesSpace发现了更多的组织亚群,但受到低质量转录物的严重影响,未能识别出具有明显空间连贯性的区域。在multi-soScope中,分辨率增强是通过从两种组学中学习的联合表示来实现的。因此,蛋白质数据也有助于生成高分辨率的转录物谱。multi-soScope增强的基因表达揭示了在大多数区域中具有更好空间连贯性的组织结构,这可以通过与原始分辨率或比较方法增强谱识别的聚类相比的聚类内距离减小来证明。仅输入蛋白质的soScope增强的蛋白质谱也提供了更精确的毛囊皮脂单位和真皮层分离的组织边界(图5c)。
接下来,作者聚焦于组织内的三个主要区域:两个毛囊皮脂亚区域和一个真皮区域。在比较不同方法的结果时,iStar和XFuse在增强RNA18S5方面表现相对较好。然而,XFuse未能生成FADS1和TETM132D的增强表达,而iStar简化了真皮层中过表达区域(图5d)。BayesSpace未能去除FADS1的噪声,也未能减少RNA18S5沿微流体阵列网格的表达模式。相比之下,multi-soScope方法有效抑制了毛囊皮脂单位外的表达水平。类似地,作者在选定区域中探索了蛋白质谱。两种方法增强的蛋白质表达准确地代表了预期的组织结构(图5e)。与原始分辨率下获得的结果相比,经过增强后,同一区域的转录物和蛋白质显示出更高的一致性,multi-soScope在Pearson相关系数方面表现最佳。通过来自不同分子类别和不同平台生成的数据集,作者证明了multi-soScope可以有效整合多组学谱,补偿质量较低的组学并共同增强多组学的分辨率。
讨论
快速发展的空间组学技术尽管实现了不同生物分子特征的空间谱分析,但仍受限于空间分辨率和数据质量。soScope是一种统一的生成框架,通过结合组学谱、空间邻近关系和形态图像,提高了多种空间组学数据的分辨率。该方法在多种技术平台上进行了广泛评估,展示了其在细化组织域识别、提高已知标记物区分度以及校正数据和技术偏差方面的有效性。soScope还可以适应空间多组学数据,支持同时增强多组学谱,并具有进一步结合单细胞组学数据以提升分辨率的潜力。
编译 | 于洲
审稿 | 曾全晨
参考资料
Li B, Bao F, Hou Y, et al. Tissue characterization at an enhanced resolution across spatial omics platforms with deep generative model[J]. Nature Communications, 2024, 15(1): 6541.
