

2024年8月19日,深势科技的Shuqi Lu、Zhifeng Gao、Linfeng Zhang和Guolin Ke研究员与北京大学的Di He教授合作,在Nature Communications上发表了一篇题为“Highly Accurate Quantum Chemical Property Prediction with Uni-Mol+”的研究成果。该研究通过开发基于深度学习的Uni-Mol+算法,创新性地利用神经网络迭代优化初始3D分子构象,精准预测量子化学属性。Uni-Mol+通过逐步逼近DFT平衡构象,显著提升了预测精度,为高通量筛选和新材料设计提供了强大的工具。

研究背景

Uni-Mol+是一种创新的深度学习方法,旨在加速量子化学属性的预测。传统方法通常依赖于1D或2D数据来直接预测量子化学属性,但这些方法在精度上往往不尽如人意,因为大多数量子化学属性依赖于经过电子结构方法(如密度泛函理论,DFT)优化后的3D平衡构象。Uni-Mol+采用了双轨Transformer模型架构,并结合了一种新颖的训练策略来优化构象生成和量子化学属性的预测。图1展示了Uni-Mol+的框架:
1. 初始3D构象生成:Uni-Mol+首先通过低成本工具(如RDKit或OpenBabel)从1D/2D数据生成初始3D分子构象。
2. 迭代更新:生成的初始构象虽然快速,但通常不够准确,因此Uni-Mol+通过神经网络进行迭代更新,使得构象逐步逼近DFT优化后的平衡构象。
3. 最终预测:在完成构象优化后,Uni-Mol+基于优化后的3D构象来预测量子化学属性,从而显著提高了预测精度。
实验结果表明,Uni-Mol+在多个公共基准数据集(如PCQM4MV2和Open Catalyst 2020)上的表现超越了所有现有的先进方法,展示了其在加速高通量筛选和新材料、新分子设计中的巨大潜力。
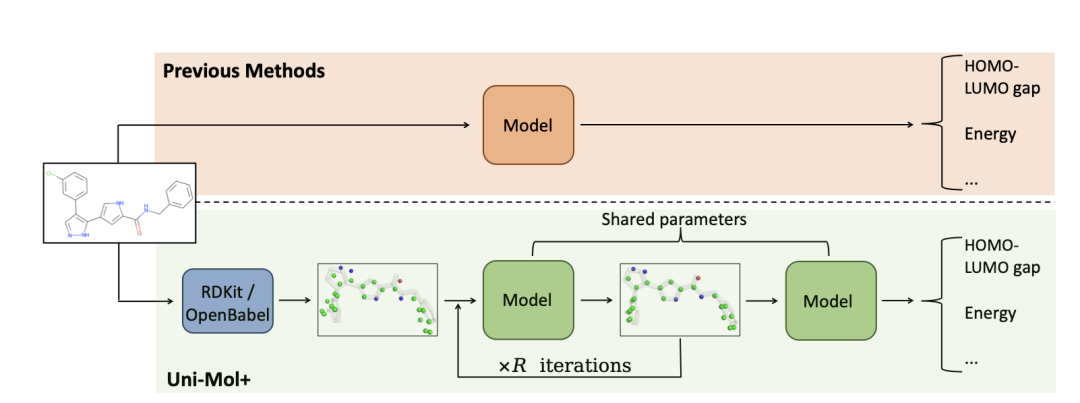
图1:与之前直接从1D/2D数据预测量子化学属性的方法不同,Uni-Mol+采用了一种不同的方式。它首先利用像RDKit这样的低成本工具从1D/2D数据生成初始3D构象,然后通过迭代更新使其逐步逼近DFT平衡构象。最终,Uni-Mol+基于优化后的构象来预测量子化学属性。

领域内的相关进展
基于1D/2D信息的深度学习模型:
早期的研究主要使用1D信息(如SMILES序列)或2D分子图来预测量子化学属性。这些方法虽然易于获取数据,但由于大多数量子化学属性依赖于经过密度泛函理论(DFT)优化后的3D平衡构象,因此预测精度往往不高。
结合3D信息的深度学习模型:
近年来,越来越多的研究开始引入3D构象信息来增强预测模型的表现。例如,有些工作在训练过程中结合了3D结构信息以提升2D表示的准确性,但推理阶段仍然主要依赖于2D信息。
3D构象优化与预测:
一些最新的研究直接从3D构象进行预测,并强调模型的旋转和平移不变性。例如,Uni-Mol通过RDKit生成的3D构象进行输入,并在预训练中引入了3D位置恢复任务。然而,这些方法在推理过程中依旧只使用了初始的3D构象,未能完全优化到DFT平衡构象。
分子构象优化的挑战:
分子构象优化是计算化学中的一个关键挑战。传统的DFT方法虽然准确但计算成本高昂。为了提高效率,提出了多种基于深度学习的潜在能量模型。这些模型通过神经网络代替DFT的高成本计算,但仍需要多步迭代来优化构象。相比之下,Uni-Mol+仅需少数几轮优化即可达到相同效果,并且能够端到端地优化构象,同时预测量子化学属性。

方法

在任何分子的处理过程中,Uni-Mol+首先使用低成本的方法(如RDKit和OpenBabel的基于模板的方法)生成初始3D构象。然后,通过从初始构象迭代更新到目标构象(即DFT优化的平衡构象)的过程,学习目标构象。最后一步,基于学习到的构象预测量子化学属性。为了实现这一目标,我们引入了一个新的模型架构和一种新的训练策略,用于更新构象和预测量子化学属性,具体内容在接下来的小节中讨论。
模型架构
研究团队设计了一个新颖的模型架构,可以同时学习分子构象并预测量子化学属性,表示为
该架构的总体设计如图2所示。与Uni-Mol相比,存在几个关键差异:
1. 位置编码:使用了原子对编码方法来编码3D空间和2D图的位置信息。具体来说,对于3D空间信息,我们使用高斯核进行编码,类似于之前的研究。此外,还引入了图的位置信息编码,如最短路径编码和一跳键编码。
2. 原子表示更新:在第l块,原子表示通过自注意力机制和前馈神经网络逐步更新。
3. 对表示更新:原子对的表示通过外积、三角更新操作和前馈神经网络进行更新。三角更新操作进一步增强了对表示的表现力。
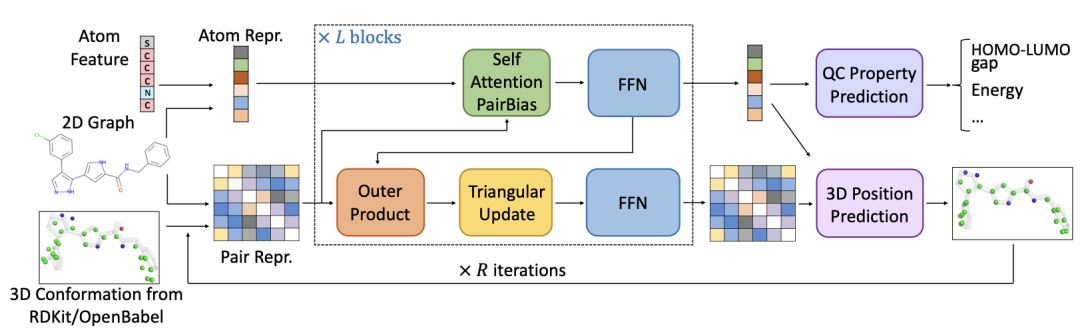
图2:Uni-Mol+的主干由多个L块组成,每个块都保持两条表示轨道——原子表示和对表示,分别由原子特征和2D图/3D构象初始化。这些表示在每个块中相互通信。基于此主干模型,Uni-Mol+通过R次迭代逐步更新初始构象(即原子的3D坐标),以接近DFT平衡构象。
模型训练
在DFT构象优化或分子动力学模拟中,构象是逐步优化的,从初始构象到平衡构象形成了一条轨迹。然而,保存这种轨迹可能非常昂贵,并且公共数据集通常只提供平衡构象。为了充分利用这些信息,我们提出了一种新的训练方法,首先生成一个伪轨迹,从中采样一个构象作为输入,以预测平衡构象。

实验验证Uni-Mol+的卓越表现
在这项研究中,研究团队对Uni-Mol+模型的性能进行了深入的实证分析。首先,团队详细介绍了Uni-Mol+的模型配置,然后利用PCQM4MV2和OC20这两个广泛认可的公共数据集,对模型在小有机分子和催化剂系统中的表现进行了全面的基准测试。通过这些测试,评估了Uni-Mol+在不同化学系统中的性能。最后,研究还通过消融实验,分析了不同模型组件和训练策略对整体性能的影响,进一步验证了Uni-Mol+的卓越表现。
模型配置
Uni-Mol+模型采用了12层结构,原子表示的维度为768,配对表示的维度为256。原子表示轨道中的FFN(前馈神经网络)隐藏层维度为768,而配对表示轨道中的FFN隐藏层维度为256。此外,在OuterProduct操作中的隐藏维度为32,TriangularUpdate中的隐藏维度也设为32。模型进行一次构象优化迭代,表明模型总共迭代两次,一次用于构象优化,一次用于量子化学属性预测。在训练策略上,随机噪声的标准差设为0.2,并使用了一种特定的采样方法来确定参数q的值。具体来说,q值为0.0的概率为0.8,q值为1.0的概率为0.1,且在[0.4, 0.6]区间内均匀采样的概率为0.1。根据这个设置,Uni-Mol+模型的参数总数大约为52.4M。
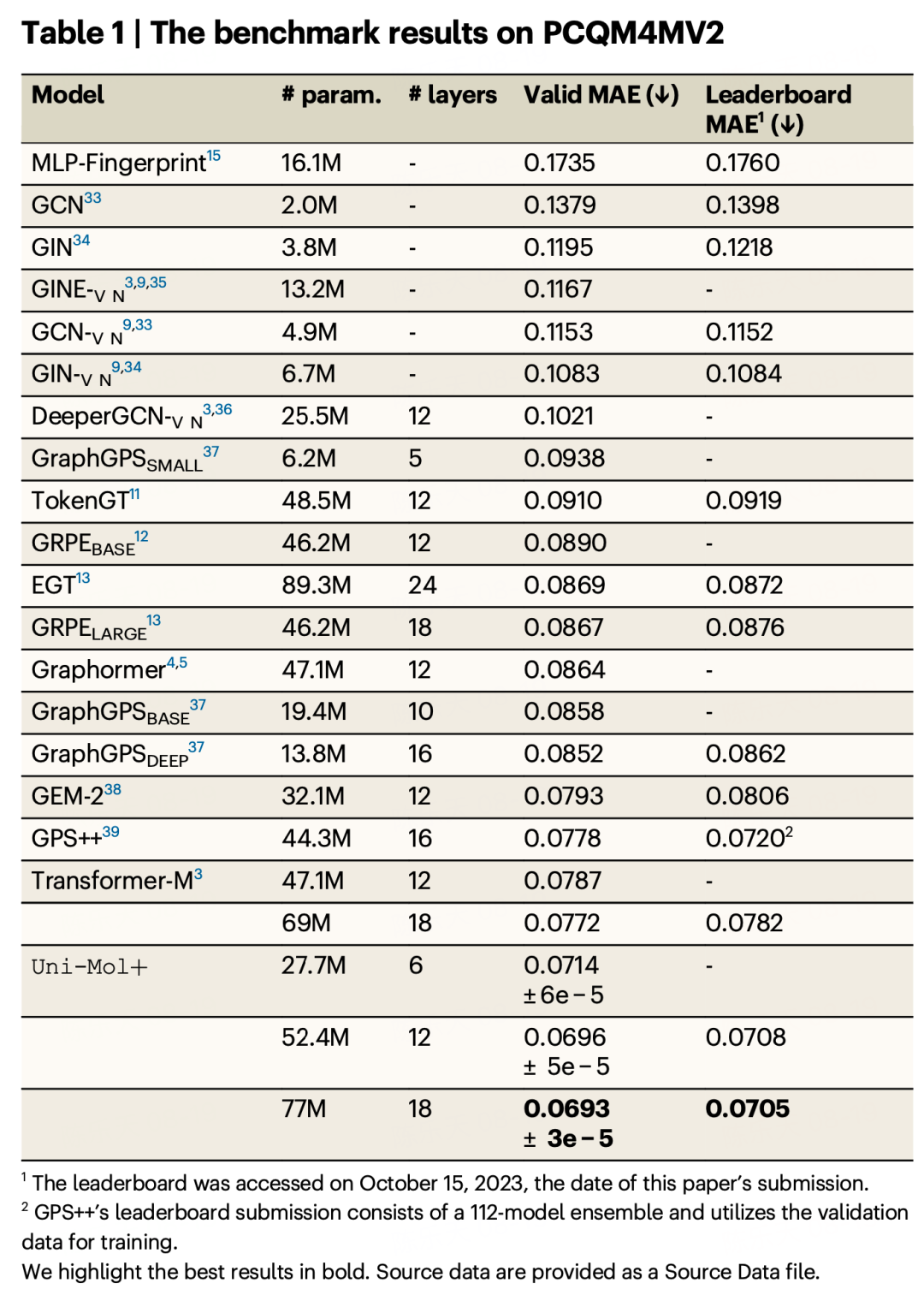
PCQM4MV2
PCQM4MV2数据集源自OGB大规模挑战赛,旨在促进机器学习模型在分子量子化学属性预测中的发展和评估,特别是目标属性“HOMO-LUMO能隙”。该属性代表了最高占据分子轨道(HOMO)与最低未占据分子轨道(LUMO)之间的能量差。数据集包含了大约400万个分子,用SMILES表示法表示。训练集和验证集提供了HOMO-LUMO能隙标签,但测试集的标签尚未公开。此外,训练集中包含了DFT优化后的平衡构象,而验证集和测试集不包含这些构象。基准测试的目标是在推理过程中仅利用SMILES表示法来预测HOMO-LUMO能隙,而不使用DFT平衡构象。
设置:基于SMILES,研究团队通过RDKit为每个分子生成8个初始构象,每个分子的生成时间大约为0.01秒。在训练过程中,在每个epoch中随机采样一个构象作为输入,而在推理过程中,使用8个构象的平均HOMO-LUMO能隙预测值。同时使用了AdamW优化器,学习率为2e-4,批次大小为1024,β1和β2设置为0.9和0.999,并且在训练过程中进行了梯度剪裁(值为5.0),训练持续了150万步,其中包括15万步的预热步骤。此外,研究团队还使用了指数移动平均(EMA),衰减率为0.999。训练过程大约持续了5天,使用了8个NVIDIA A100 GPU。在147k的测试开发集上进行推理时,使用了8个NVIDIA V100 GPU,大约耗时7分钟。
研究团队将之前在PCQM4MV2排行榜上的提交结果作为基线。除了默认的12层模型外,我们还评估了Uni-Mol+的两个变体,即6层和18层模型,以研究不同模型参数规模对性能的影响。
结果:结果总结在表1中,要点归纳如下:
1. 所有三个变体的Uni-Mol+都在之前的基线方法上表现出显著的性能提升。
2. 即使是6层的Uni-Mol+模型,尽管其参数量较少,仍然超过了所有之前的基线方法。
3. 将层数从6增加到12后,精度显著提升,远超所有基线方法。
4. 18层的Uni-Mol+模型表现最佳,在所有基线方法中取得了显著优势。
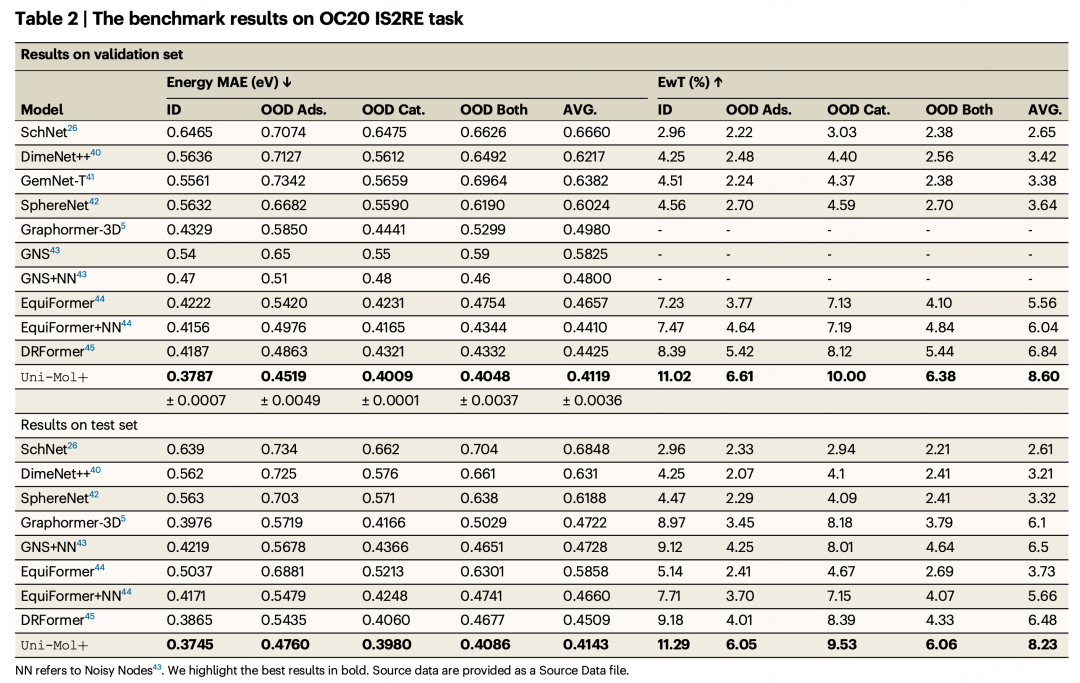
Open Catalyst 2020
Open Catalyst 2020 (OC20)数据集旨在推动机器学习模型在催化剂发现和优化方面的发展。OC20包括三个任务:从结构到能量和力(S2EF)、从初始结构到放松结构(IS2RS)以及从初始结构到放松能量(IS2RE)。本文主要关注与研究团队提出的方法目标相符的IS2RE任务,该任务的目标是基于初始构象预测放松能量。该数据集包含大约46万个训练数据点。虽然DFT平衡构象在训练中提供,但在推理过程中禁止使用。此外,与PCQM4MV2数据集不同,OC20的IS2RE任务已经提供了初始构象,因此无需自行生成初始输入构象。
设置:研究团队在OC20实验中使用了默认的12层Uni-Mol+配置。与 PCQM4MV2的设置略有不同,具体如下:首先,由于OC20数据集缺少图信息,从模型中排除了与图相关的特征。其次,由于OC20数据集中的原子数量较PCQM4MV2多,为了提高效率,模型容量略有降低。具体来说,配对表示维度设置为128,而OuterProduct和TriangularUpdate中的隐藏维度则分别设置为16。第三,由于需要考虑周期性边界条件,研究团队采用了一种预先扩展邻居单元并应用半径截断的方法来减少原子数量。训练过程中使用了AdamW优化器,训练持续了150万步,包括15万步的预热步骤。优化器的学习率设置为2e-4,批次大小为64,β1和β2的值分别为0.9和0.999,梯度剪裁参数为5.0。整个训练过程大约持续了7天,使用了16个NVIDIA A100 GPU。
结果:表2和表3中展示了各种模型在OC20 IS2RE验证集和测试集上的性能比较。两张表中显示了各模型的能量平均绝对误差(MAE)和符合阈值的能量百分比(EwT%)。从表中可以看出,研究团队提出的Uni-Mol+在MAE和EwT两个方面都显著优于之前的所有基线方法,表现出卓越的性能。特别值得注意的是,Uni-Mol+方法在所有类别中都取得了最低的MAE值,包括域内(ID)、域外吸附(OOD Ads.)、域外催化(OOD Cat.)、域外综合(OOD Both)和平均(AVG)分类。此外,在EwT指标上,Uni-Mol+也在所有类别中取得了最高的得分。这些结果强调了Uni-Mol+在处理域内和域外数据方面的稳健性,并展示了其在材料系统中捕捉复杂相互作用的有效性,以及在广泛的计算材料科学任务中的潜在应用。

总结

在这篇研究中,Uni-Mol+模型通过深度学习为量子化学属性预测开辟了一条新的路径。与传统方法不同,Uni-Mol+首先利用低成本的工具从1D/2D数据生成初始3D构象,然后通过神经网络逐步将其优化至DFT平衡构象,再基于优化后的构象进行量子化学属性的预测。研究结果表明,Uni-Mol+在多个公共基准数据集(如PCQM4MV2和Open Catalyst 2020)上的表现显著优于此前的所有方法。消融研究进一步验证了模型各组成部分的有效性。总体而言,Uni-Mol+展示了通过深度学习加速量子化学属性预测的巨大潜力,为未来的高通量筛选和新材料、新分子设计提供了有效的工具。

