
DRUGAI
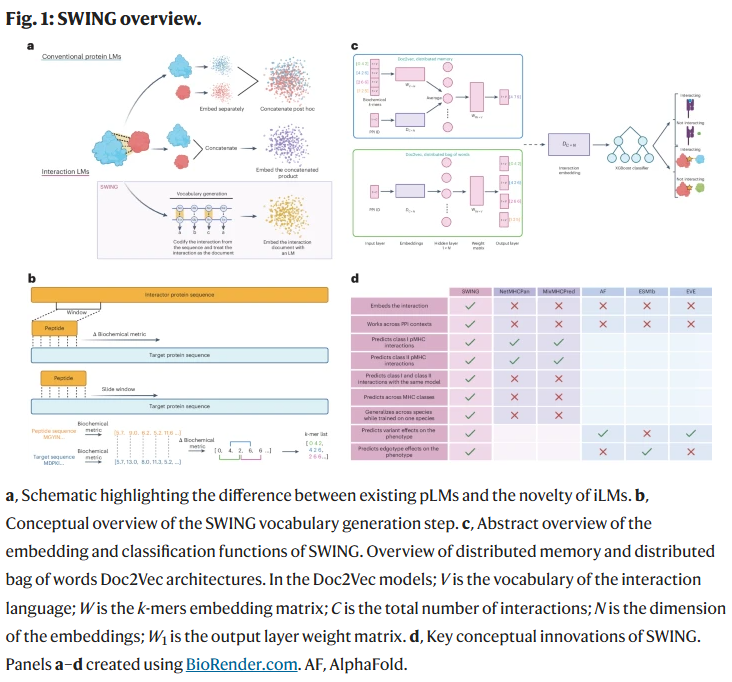
尽管当前的蛋白质语言模型(protein language models, pLMs)在序列嵌入任务中表现出色,但它们在解析和建模蛋白质间互作语言方面仍存在明显局限。为此,研究人员提出了SWING(Sliding Window Interaction Grammar)——一种新型的交互语言模型(interaction language model, iLM),该模型通过滑动窗口机制捕捉氨基酸性质的局部差异,生成一种具有生物意义的“互作词汇”,并据此构建序列层面的交互表达。
SWING不仅能够准确预测MHC-I与MHC-II类的肽段-主要组织相容性复合物(pMHC)结合,还具备跨类别泛化能力,首次实现了用MHC-I模型跨预测MHC-II互作。此外,SWING在人类训练集上学习到的模型可直接迁移至小鼠MHC-II互作预测,覆盖系统性红斑狼疮(SLE)和1型糖尿病等免疫相关疾病风险等位基因。研究人员进一步表明,SWING可识别由错义突变引发的蛋白-蛋白互作扰动,且无需依赖结构信息。整体而言,SWING是一种具备零样本预测能力、可广泛应用于多种蛋白互作建模场景的通用性iLM。

深度学习方法已彻底革新了分子预测与生成任务,特别是在基因组学和蛋白组学中,生成模型的应用推动了功能蛋白设计和免疫识别机制的深入研究。近年来,pLMs成为理解和工程化蛋白序列的重要工具,可用于设计类似天然蛋白功能的合成序列,或建模抗体进化过程等。然而,当前pLMs普遍采用将蛋白序列分别编码再合并的方法进行蛋白互作预测,这种方式忽略了真正起作用的互作界面残基之间的语义联系。
蛋白质的功能通常通过与其他蛋白质的互作实现,但这种互作行为受到严格的进化约束和结构规律控制。现有pLM方法在生成整体序列嵌入时,难以提取互作残基对之间的生化特征,也难以适应不同序列长度、不同物种或未知等位基因等广泛变异情境。因此,研究人员开发SWING框架,重新定义互作的表示方式,从语言学角度捕捉蛋白-肽互作背后的语法规则,从而实现通用、灵活、可迁移的预测能力。
结果
SWING框架与设计原理
SWING的核心思想是将蛋白质互作看作一种“语言行为”,每对氨基酸之间的生化属性差异构成“词汇”,而肽-蛋白或蛋白-蛋白之间的互作序列则构成“句子”或“文档”。该模型通过滑动窗口策略扫描互作序列对,在每个位置计算生化指标(如极性、疏水性)差异,构建有序序列向量,进而生成嵌入。SWING利用Doc2Vec进行无监督嵌入训练,再将嵌入输入至XGBoost等监督学习模型中完成预测任务。该预嵌入方式相比传统pLM方法具有更强的互作上下文表达能力,无需对现有语言模型结构进行修改或微调。
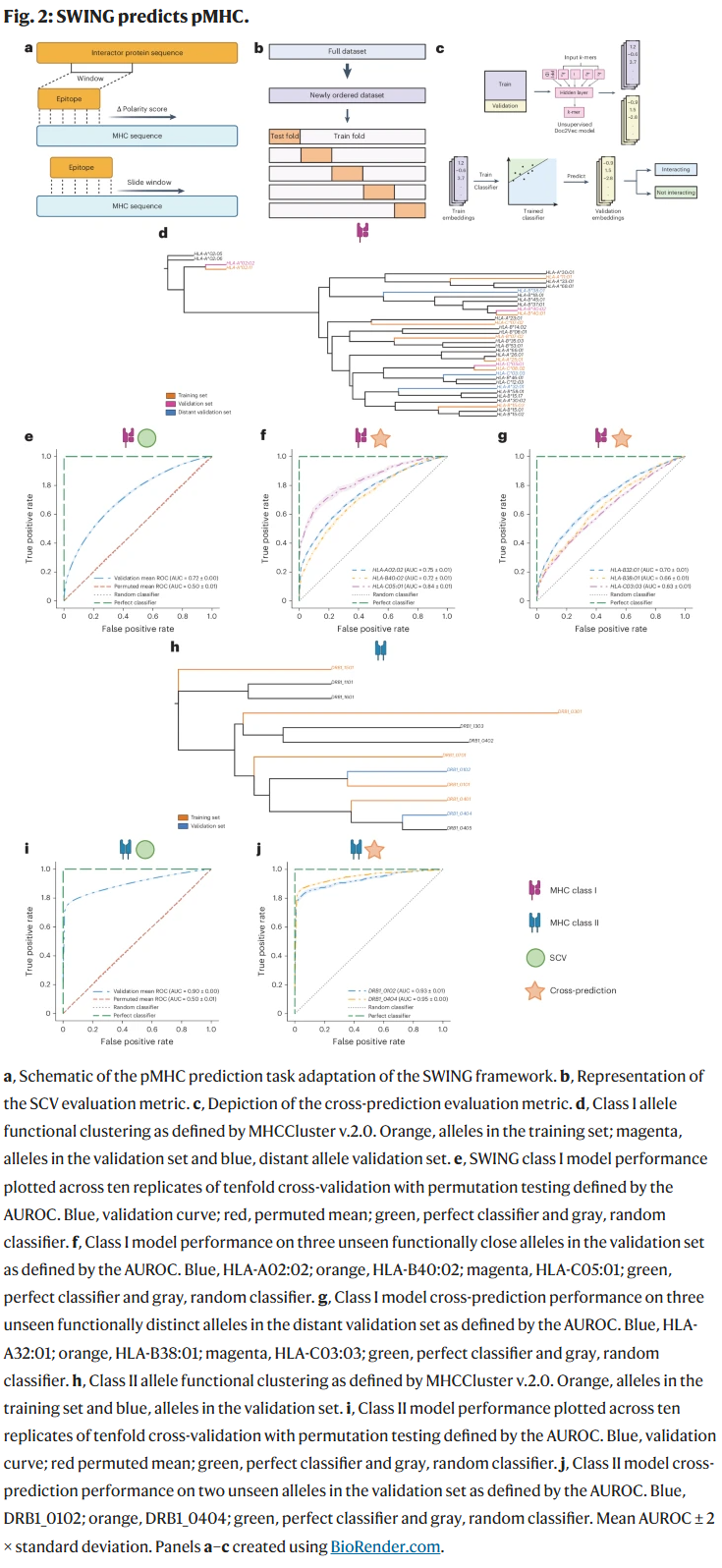
SWING精准学习pMHC互作语言
在pMHC预测任务中,研究人员分别对MHC-I和MHC-II肽段互作进行了建模,发现SWING能够准确识别功能上互补但结构差异显著的互作机制。在10折交叉验证及跨等位基因预测中,SWING在不同HLA等位基因上的AUC范围为0.72–0.95,表现稳定,优于多数现有方法。
更重要的是,SWING-MHC-I模型具备预测MHC-II互作的能力,且在预测未曾见过的等位基因上,保持良好的泛化性能。该跨类别预测能力说明SWING模型中嵌入的互作语言具有生物学普适性。
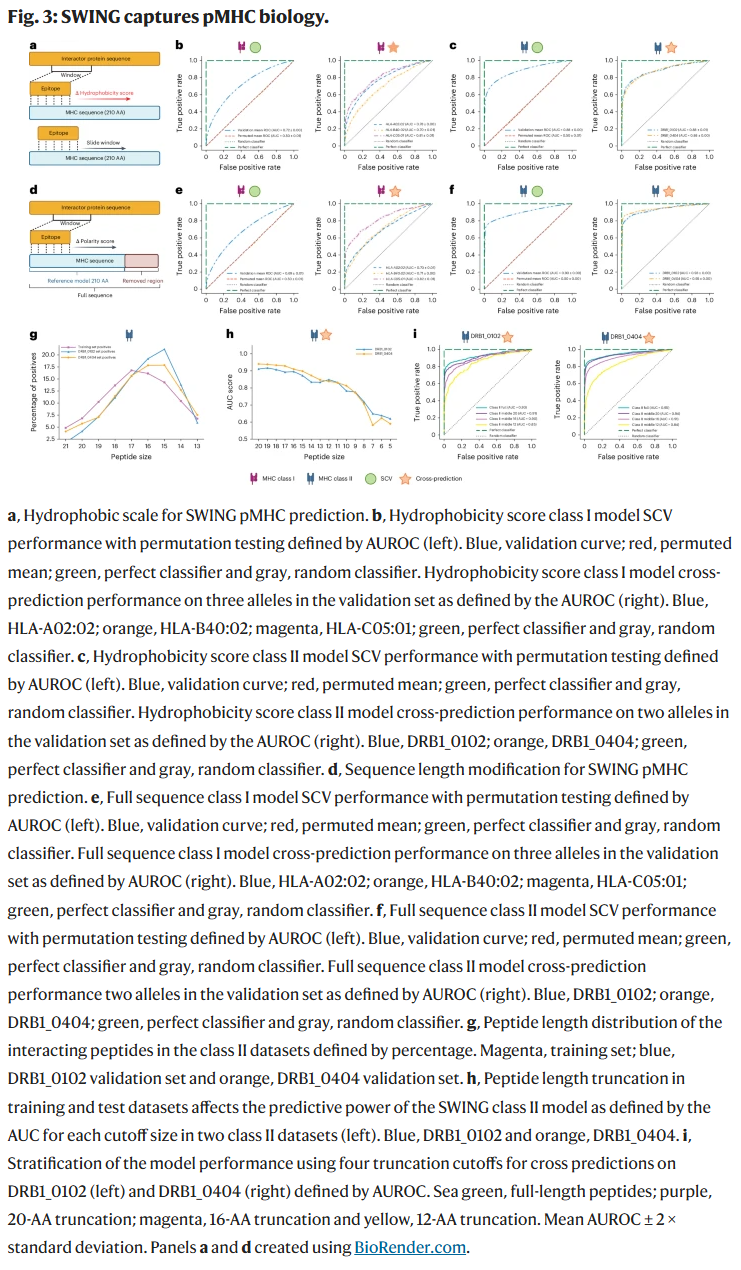
SWING捕捉互作中的生物学信息
研究人员进一步分析了SWING对生物学关键特征的敏感性。当将滑动窗口中计算的生化指标从极性替换为疏水性时,模型性能仍保持一致,说明SWING建构的交互语言不仅稳健而且具备可解释性。此外,模型对肽段长度表现出高度依赖性,长度小于9个氨基酸时性能大幅下降,验证了SWING能自动捕捉pMHC中“核心结合基序”的重要性,而无需明确指定。
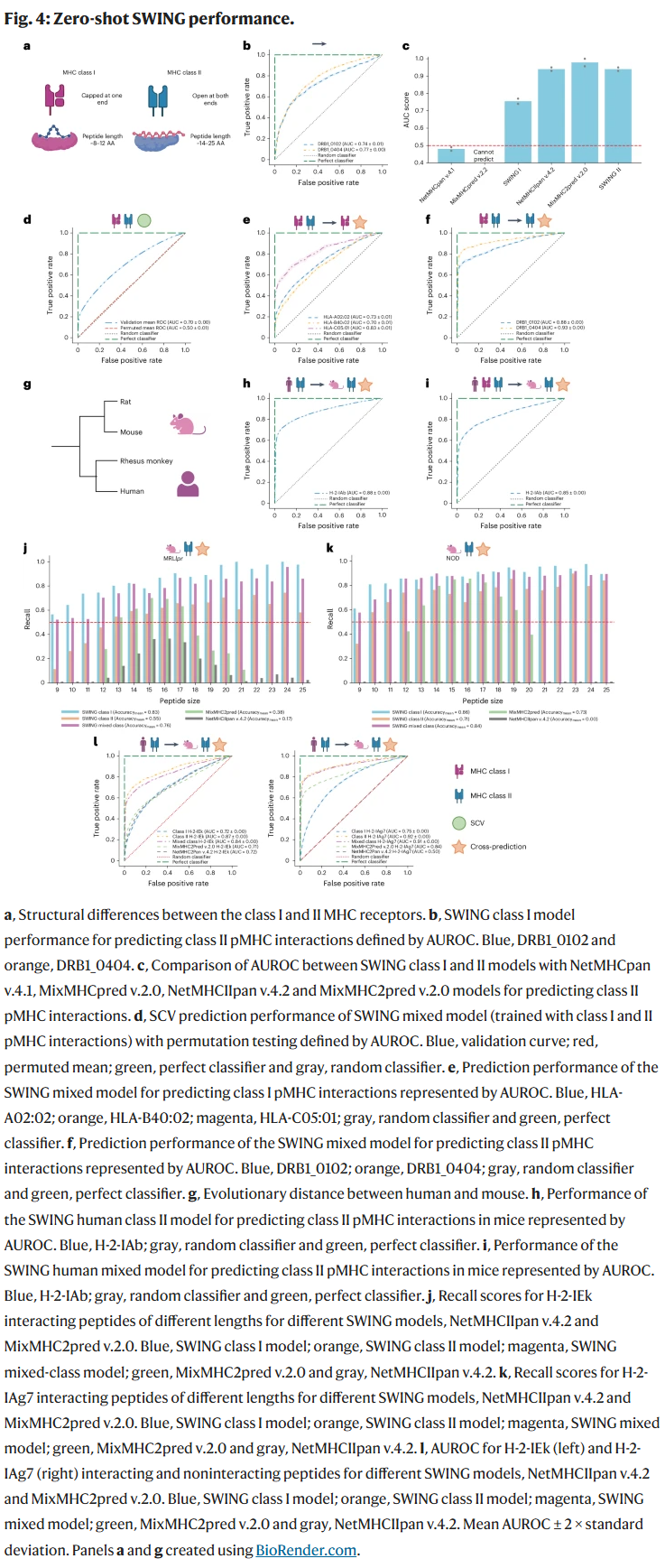
SWING具备跨任务、跨物种、跨类别迁移能力
研究人员构建了联合训练模型(覆盖MHC-I与MHC-II数据),不仅提升预测性能,也使模型在新任务上具有“零样本”迁移能力。以人类训练的模型为例,SWING在小鼠的H-2-IEk与H-2-IAg7等位基因上实现高准确率(AUC达0.85–0.92),优于现有NetMHCIIpan与MixMHC2pred模型,后者虽包含目标等位基因但预测表现更差。SWING甚至在肽段长度不在训练集中(如22–36 AA)仍表现稳定,证明其在免疫识别中的广泛适应性。
SWING预测蛋白互作扰动
研究人员进一步扩展SWING用于预测由错义突变引发的蛋白-蛋白互作扰动,不依赖三维结构信息。模型仅通过序列中滑动窗口所包含的突变残基和其互作伙伴残基对之间的生化语言特征,即可高效识别哪些变异可能破坏互作。无论是孟德尔遗传病变异还是常见群体变异,SWING均可有效预测其引发的互作破坏(AUC高达0.87)。值得注意的是,SWING甚至能够识别界面外突变引发的构象改变所导致的功能丧失。
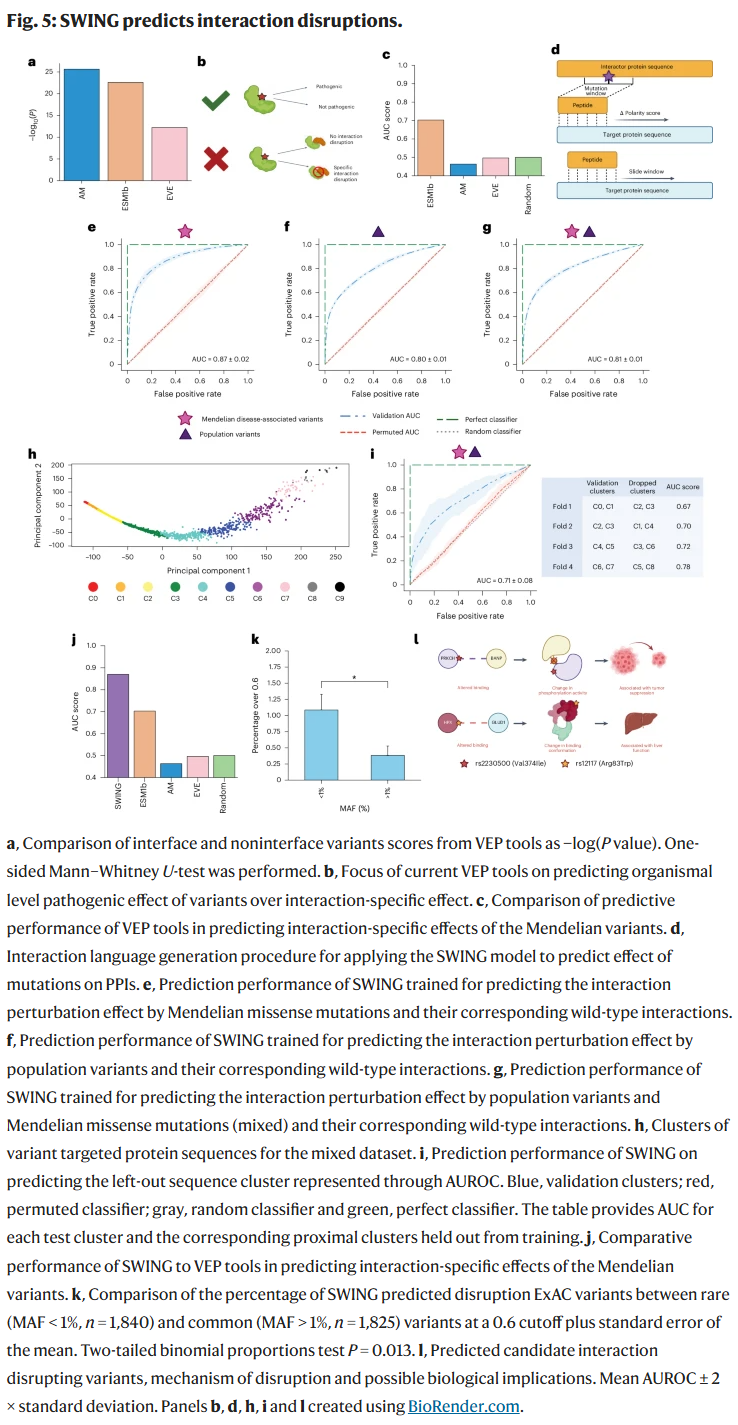
SWING超越传统VEP工具
当前主流的变异效应预测工具(如AlphaMissense、EVE、ESM1b)虽在整体致病性分类上表现良好,但缺乏对“特定互作扰动”的解析能力。SWING通过建模“交互语言”,提供了比常规结构或保守性方法更细粒度的预测能力。在不同模型架构下(如CNN、Doc2Vec、XGBoost等),SWING的嵌入均展现出强鲁棒性与迁移能力,表明其语言本身足够表达互作语义,而不仅仅依赖特定模型。
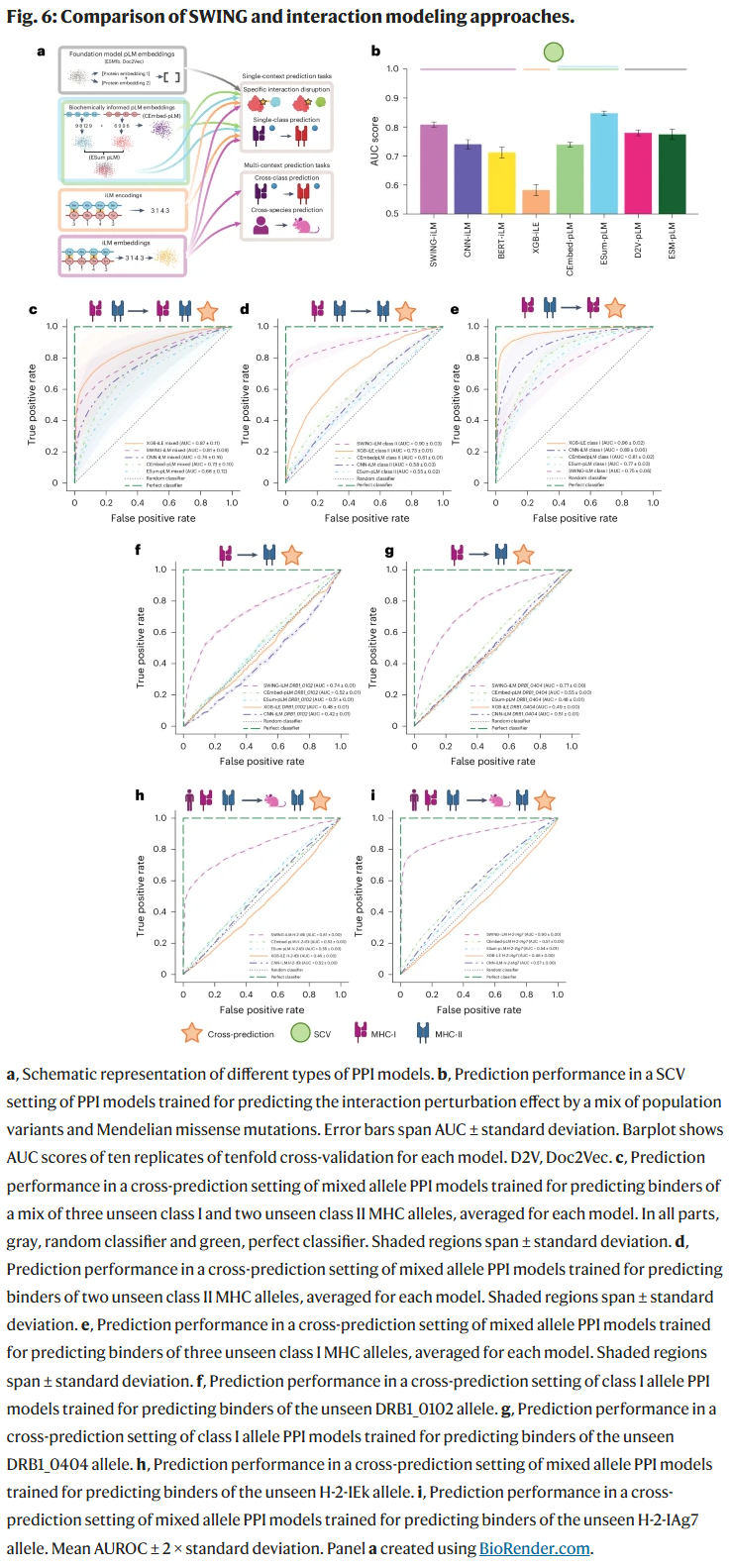
讨论
SWING代表了一种全新的蛋白质互作建模范式,其生物启发式设计和交互语言表达方式,使其具备出色的泛化能力与跨领域适用性。相较于现有pLMs将蛋白质作为独立实体建模的策略,SWING将互作本身作为建模目标,通过滑动窗口提取局部生化“语法”,建立起一套结构与功能兼容的交互语言体系。
无论是在免疫相关肽段识别、跨物种互作预测,还是错义突变引发的互作扰动分析中,SWING均展现出强大的预测能力和解释性。其零样本学习能力使其尤为适合应用于稀有等位基因、长肽段或未知变异的研究情境中。未来,SWING可进一步集成结构、动力学、表达等多模态信息,构建更全面的蛋白互作预测平台。
整理 | WJM
参考资料
Siwek, J.C., Omelchenko, A.A., Chhibbar, P. et al. Sliding Window Interaction Grammar (SWING): a generalized interaction language model for peptide and protein interactions. Nat Methods (2025).
https://doi.org/10.1038/s41592-025-02723-1
内容中包含的图片若涉及版权问题,请及时与我们联系删除
