
DRUGAI
目前最常用、预测能力最强的蛋白质模拟模型是全原子分子动力学方法,但其计算代价极高。开发一种预测能力相当且具备广泛适用性与高计算效率的粗粒度(coarse-grained, CG)模型,一直是该领域的长期挑战。研究人员在本研究中结合了最新的深度学习方法与大规模多样化的全原子蛋白质模拟数据,开发出一种具有化学可转移性的自底向上的CG力场。该模型可用于对未参与参数训练的新蛋白质序列进行外推模拟。研究人员展示了该模型在多个方面的预测能力:包括折叠、解折叠、中间构象的亚稳态,内在无序蛋白的波动行为,以及突变体之间的相对自由能差异,同时其计算速度比全原子模型快几个数量级。这表明,开发一种普适的、计算效率高的、基于机器学习的蛋白质CG模型是可行的。

过去几十年,随着硬件性能提升、模拟软件的进步以及理论方法的发展,蛋白质等大分子的分子模拟已从最初的概念验证阶段发展为可深入研究其折叠过程与构象动力学的重要工具。然而,如何在保证精度的同时兼顾效率,仍是当前面临的核心挑战。
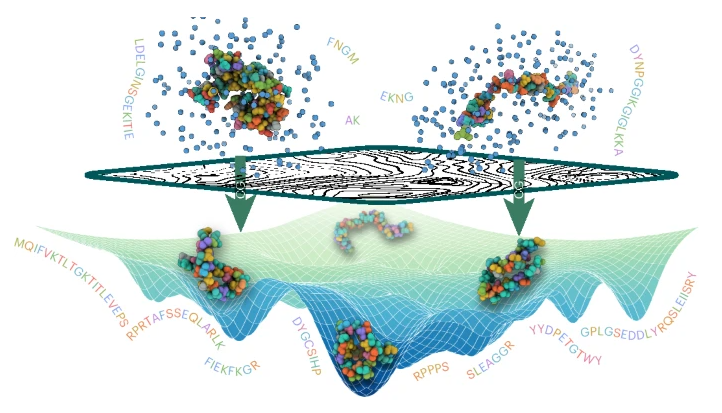
蛋白质系统具有多尺度的动态特征:局部、快速的原子层级变化需要高分辨率描述,而整体、长时间尺度的动力学过程则更适合在粗粒度层面进行建模。粗粒度模型通过减少自由度,使得计算更高效,并便于理解微观相互作用如何导致宏观行为的产生。尽管如此,目前使用最广泛、最精确的模拟方式仍然是基于全原子模型的分子动力学,其计算资源消耗极大,且需要复杂的后处理分析。
另一方面,深度学习方法近年来在蛋白质结构与功能预测方面展现了惊人的能力,尤其是在大规模数据集驱动下。但这些方法往往缺乏对物理机制的建模能力,难以应用于解析蛋白质热力学过程与动力学演化。
研究人员提出了一种新的思路:使用深度神经网络学习全原子模拟中的构象分布,从而构建出一套具有物理意义的、多体相互作用驱动的粗粒度力场模型。该模型不仅能用于蛋白质结构预测,更关键的是能模拟其构象转变过程,自主学习亚稳态之间的转化机制,并具备较强的外推能力。
结果
构建训练数据与模型开发
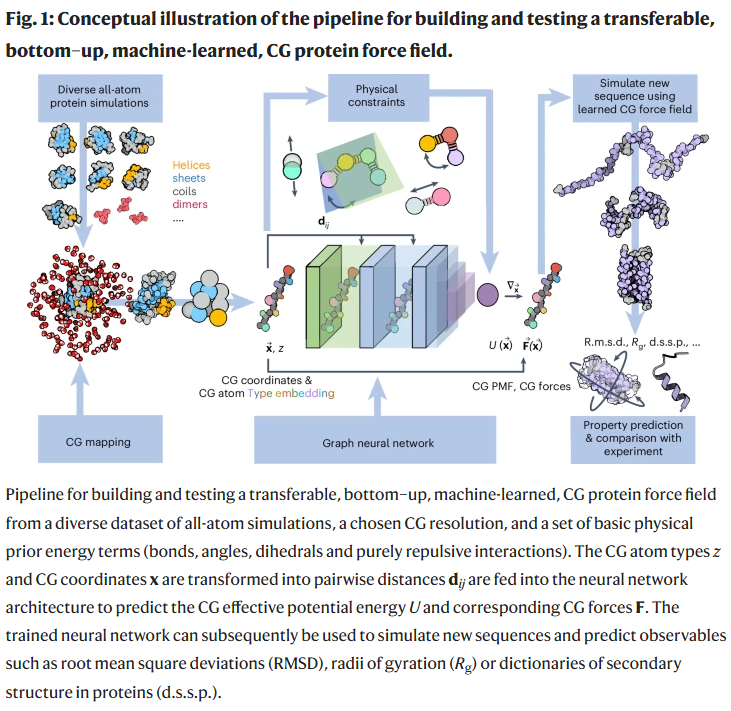
研究人员建立了一个大规模的训练数据集,包括50种不同折叠类型的小型蛋白质以及超过1200个肽二聚体的全原子显式溶剂模拟。结合图神经网络架构与物理先验能量项,训练出可转移的粗粒度力场CGSchNet,并在多个未见过的蛋白质上开展自由能景观重建与动力学模拟实验。
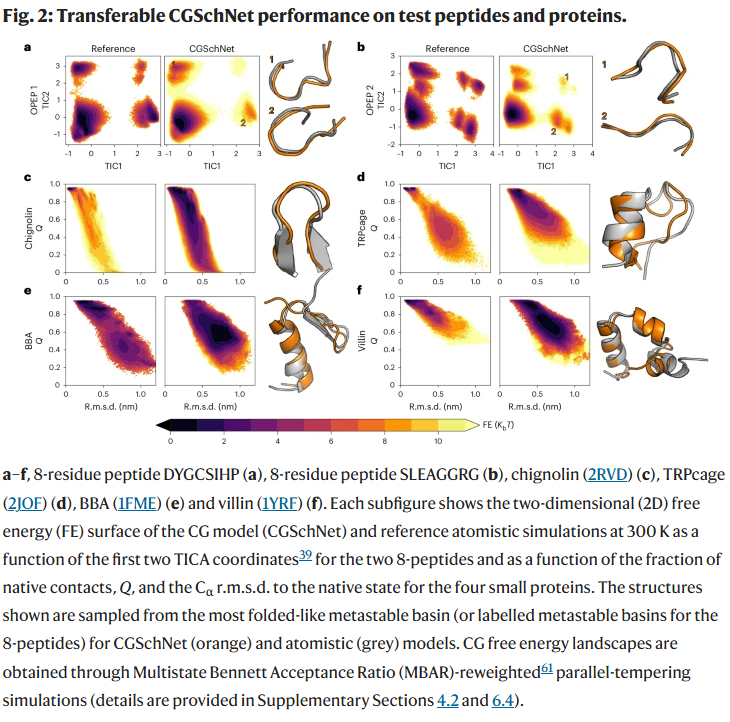
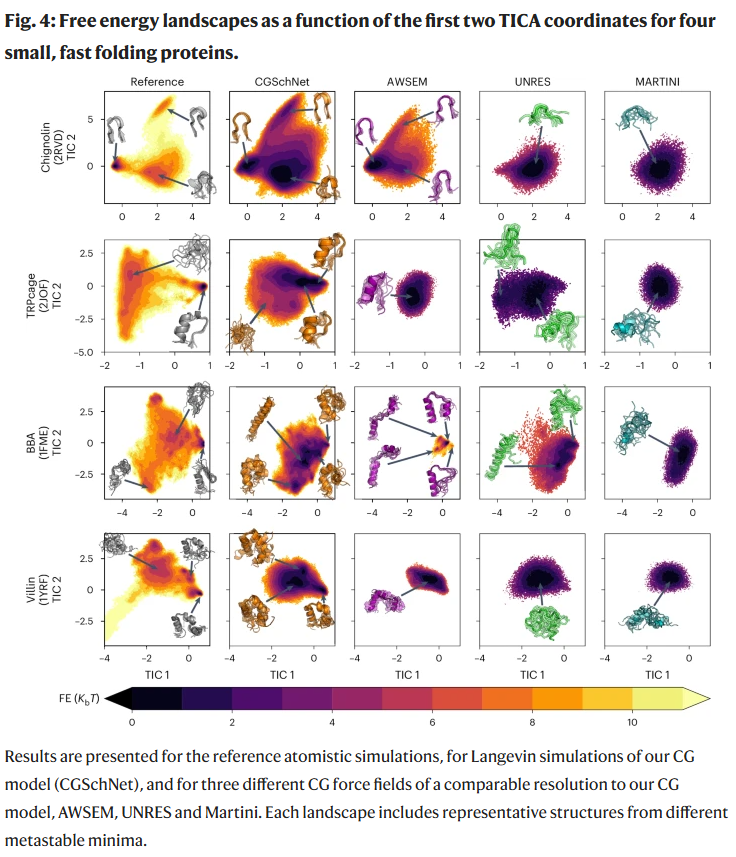
小型肽与快速折叠蛋白的自由能景观预测
在未参与训练的8残基肽(如DYGCSIHP、SLEAGGRG)及快速折叠蛋白(如Chignolin、TRPcage、BBA、Villin)上,CGSchNet预测出的自由能景观与全原子模拟结果高度一致。对于小肽而言,构象主要由扭转角度决定,因此神经网络的作用相对较小;而对于蛋白质而言,模型成功捕捉了折叠与解折之间的转变状态,并预测出正确的构象亚稳态。这些折叠构象在天然接触比(Q值)与Cα主链RMSD等指标上接近于晶体结构,验证了模型在蛋白质构象预测上的高准确性。
值得注意的是,模型不仅能再现天然折叠态,还能识别出类似于Chignolin中的错误折叠状态(如TYR1与TYR2错配),这在以往的CG模型中较为罕见。
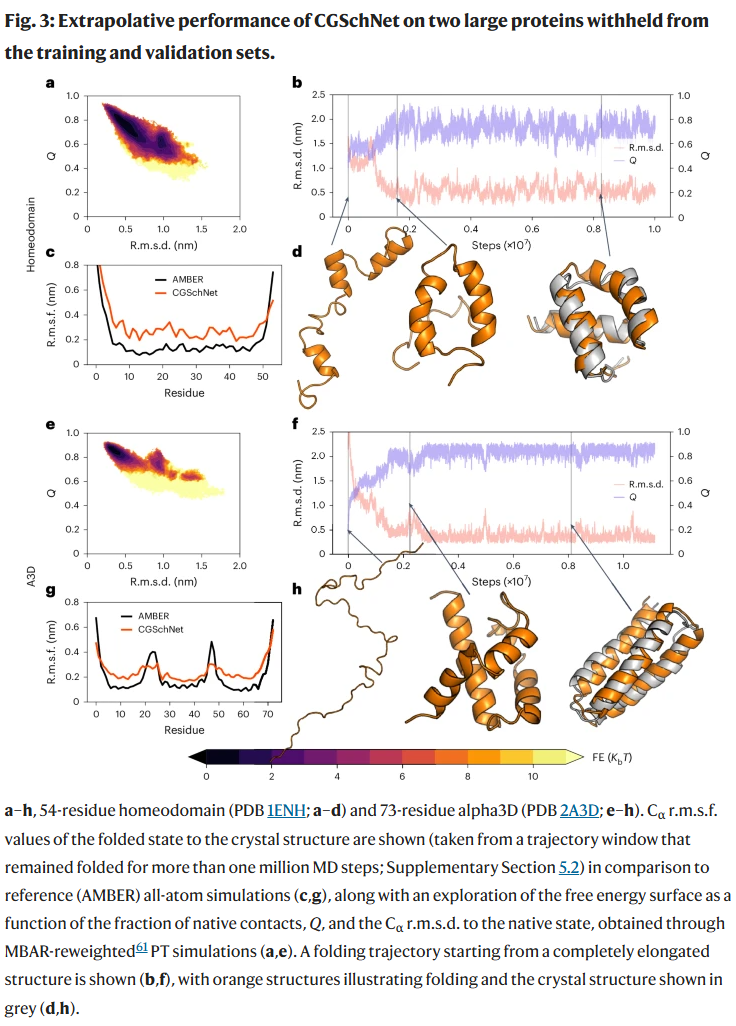
外推至更大蛋白质
研究人员将模型应用于更大、结构更复杂的蛋白质,如homeodomain(54残基)与de novo设计的alpha3D(73残基),评估其在无训练数据支持下的泛化能力。结果显示,CGSchNet能从拉伸构象中自动折叠出正确的天然构象,并预测出与参考晶体结构一致的主链波动。特别是在alpha3D中,模型还能识别出与天然结构仅有轻微差异的三螺旋替代拓扑结构,表明其具备一定的构象多样性识别能力。
与主流CG力场的系统性对比
研究人员将CGSchNet与三种常用CG模型(AWSEM、UNRES、Martini)进行了全面对比。在快速折叠蛋白(如Chignolin、TRPcage、BBA、Villin)上的TICA坐标自由能图中,CGSchNet能够分辨多个构象态(折叠、未折叠、错误折叠等),而其他模型往往只稳定一个主态甚至无法正确预测折叠。特别是在BBA中,该蛋白质含有α-螺旋与β-折叠混合结构,是CG建模的难点,CGSchNet依然能识别出局部亚稳折叠态,而其他模型基本失败。
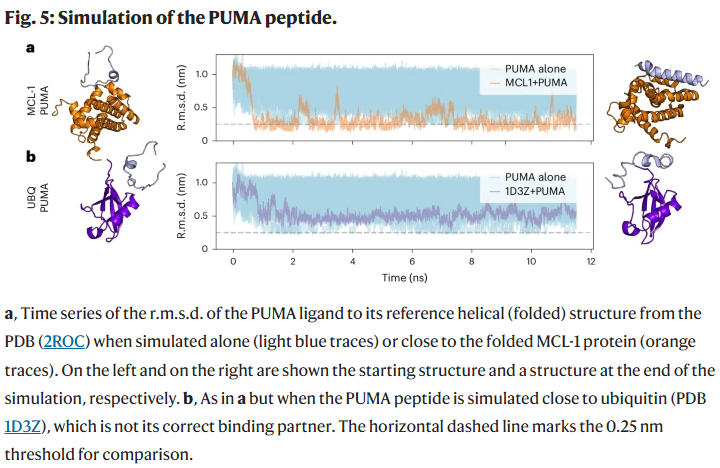
无序肽在结合中的折叠行为预测
研究人员以PUMA-MCL-1体系为例,考察模型是否能外推预测“无序肽结合后折叠”的行为。在无结合蛋白时,PUMA肽处于高度波动、无折叠状态;而在存在MCL-1时,肽迅速形成稳定的α螺旋结构,RMSD快速下降至2.5 Å以下,表明模型成功捕捉了配体诱导折叠机制。作为对照,与非结合蛋白ubiquitin共模拟时,PUMA未出现稳定折叠,进一步验证了模型的特异性。
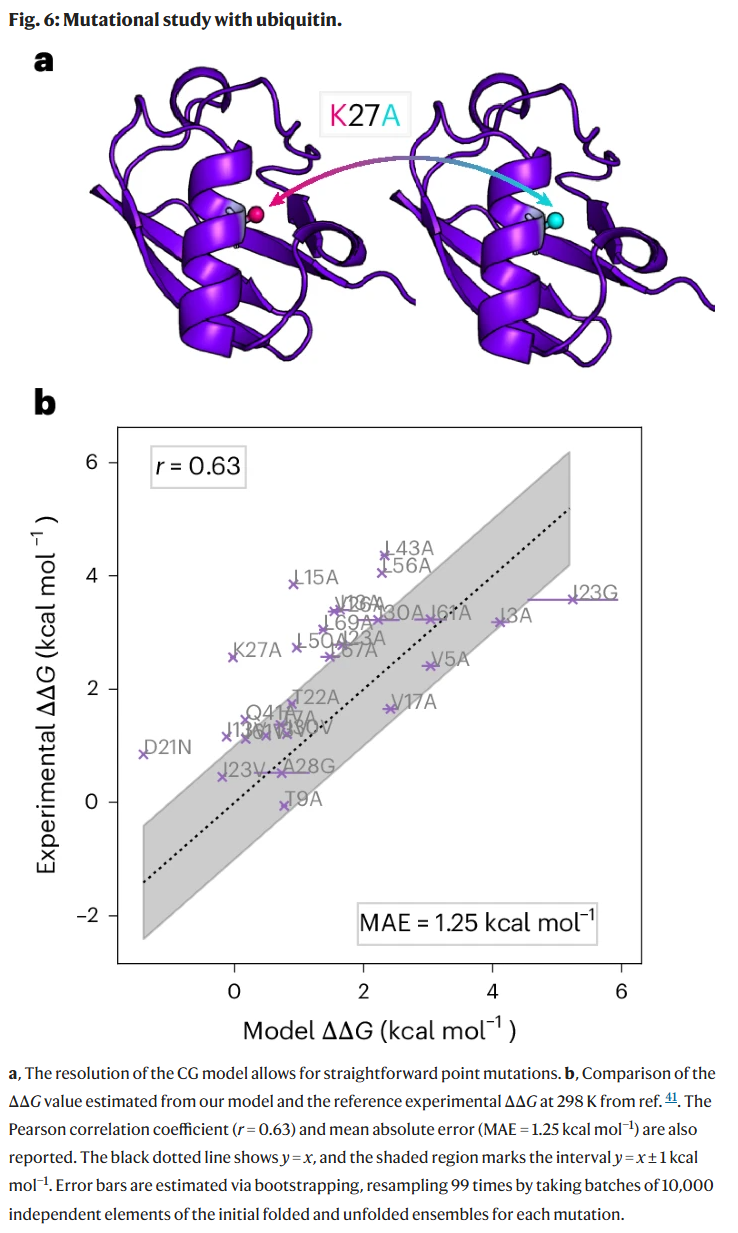
蛋白突变的自由能变化预测
研究人员还使用CG模型评估了ubiquitin蛋白在一系列点突变下的折叠自由能变化,并与实验ΔΔG数据进行了比较。结果显示,两者之间具有良好的线性相关性,表明该模型不仅具备构象预测能力,还可用于蛋白稳定性评估与突变效应分析。
讨论
研究人员提出的CG模型CGSchNet,结合图神经网络的表达能力与自底向上的多体相互作用建模方式,在较小训练数据集的前提下,实现了强大的转移能力与预测性能。模型不仅可用于小型蛋白质的折叠研究,也能外推至更大的体系,并具备对非天然结构、突变体与配体结合事件的敏感性与分辨力。
尽管该模型并非以结构预测为主,但它提供了一个探索蛋白质自由能景观、识别多构象状态、揭示折叠/解折过程、预测突变效应与配体诱导折叠机制的强大平台。相比之下,AlphaFold等方法主要集中于稳定结构预测,忽视了蛋白质在生理环境中的多态性与动力学行为。
研究人员指出,尽管当前模型尚未考虑温度与压力等环境条件的变化,未来可通过加入条件建模机制、拓展训练数据至多温度多体系来进一步增强模型泛化能力。此外,模型的先验能量项对其物理稳定性起到关键作用,需进一步系统优化;在分辨率选择、长程作用建模与网络架构改进方面也存在提升空间。
总的来看,本研究展示了如何将深度学习与物理建模相结合,实现一种通用、可解释且高效的蛋白质模拟工具。这一成果为未来开发具备预测能力的粗粒度模型打下了坚实基础,并为蛋白质动力学、突变筛选与分子设计等任务提供了新方向。
整理 | WJM
参考资料
Charron, N.E., Bonneau, K., Pasos-Trejo, A.S. et al. Navigating protein landscapes with a machine-learned transferable coarse-grained model. Nat. Chem. (2025).
https://doi.org/10.1038/s41557-025-01874-0
内容中包含的图片若涉及版权问题,请及时与我们联系删除
