
Published on July 28, 2025 9:13 PM GMT
There is no consensus in aging research on how to define, measure, or explain aging, with fundamental disagreements over its causes, onset, and reversibility.
Examples of the diverse mechanistic definitions of aging.
“We do not know what causes aging. I think the trigger is endogenous but not necessarily programmed.” (G. Ferbeyre)
“Aging is mainly accumulation of macromolecular damage and failure of adaptation to a continuous changing environment. It is also an epiphenomenon of the outcome of the protective conditions we humans have achieved thanks to civilization over time and to this end somehow it doesn't comply fully with typical Darwinian Biology.” (E.S. Gonos)
“As an evolutionary biologist, I see 'aging' as the decrease in the age-specific contribution to fitness. […] At a mechanistic level, aging (or I should say 'senescence') corresponds to any deterioration of cellular/physiological traits that will ultimately impact fitness.” (J.F. Lemaître)
“Mechanistically, aging is a disruption of the homeostasis established between cellular processes. Age-related-diseases are the external symptoms of this disruption. […] Aging is the result of [an] imperfect optimization to maintain a balance between evolutionary constraints, cellular dynamical equilibrium and environmental constraints. […] Aging is not a programmed process but a consequence of this search for an equilibrium.” (Q. Vanhaelen)
“Time-dependent degradation of interactions or their (quasi)adaptative changes; sluggish and/or ineffective responses to challenges; when more than one such response is biologically relevant, aging would manifest as preference for a response different than one preferred at youth. The latter response may still be effective, but at higher cost for an aging organism (e.g. increased innate, inflammatory responses to pathogens in the old).” (J.M. Witkowski)
“Aging is the progressive decline of function with increasing chronological age due to internal factors that are not dependent on environment, which leads to an increased probability of death. Aging is caused by a genetically programmed switch that downregulates cellular pathways involved in homeostasis, stress response, repair etc. that evolved to limit competition for resources between offspring and parent.” (J. Van Raamsdonk)
“I believe that some mechanism that clears damage from the germline (which is immortal) is toxic to the rest of the organism. This toxicity interferes with transcription at the DNA or protein level, leading to imbalanced proteostasis. Chromatin condensation therefore becomes abnormal (as measured by epigenetic clocks) which disrupts function of the organism as a whole and likely exponentially through a negative feedback loop.” (T. Liontis)
“Ageing is caused by a breakdown in repair mechanisms due to a shift of resource allocation after reproduction, modulated according to the environmental niche of the organism.” (G. Pawelec)
“Aging is not programmed. Aging is the result of complex interactions between the genome and physiological and environmental changes, with feedback loops that lead to physical and mental impairment. Organisms are not adapted to naturally live indefinitely.” (F. Dufour)
“Repair machineries fail to keep up with internal and external damage; systems become dysregulated and eventually collapse.” (V. Gorbunova)
“The gradual break-down of cellular components, leading to the eventual death of the organism, associated with time. This is caused by DNA damage, mutations, genetic/epigenetic pre-programmed senescence, protein aggregates and environmental stress.” (U. Anglas)
A disconnection in the literature exists between mechanistic (molecular) and evolutionary models of aging. Here i will try to bridge that gap by taking the core ideas of Kirkwood´s disposable soma theory of aging and Sinclair´s information theory of aging and propose a model to answer open questions over the mechanisms and reversibility of aging. With that i want to establish a better definition of aging research. At the moment, basically anything can be considered aging research as long as you examine the changes in a body part over time.
I am a mechatronics engineer who turned to aging research. This post is not highly polished, I would be surprised if nothing i write here is wrong and it could definitely use some more references for the claims i make as well as better introductions for specific biology concepts. I will link to explanations where I think they are useful for people not coming from this field. At certain points, I’ll include any doubts I have about my own writing in <details>, along with possible directions from where they could be addressed.
Contents
In "1. Adaptation: Nature’s Strategy for Survival and Success" I explain concepts that are at the core of nature. It is my understanding of what nature is trying to optimize. I will also explain some examples that prove my point.
In "2. Model for Adaptive Gain" I take that concept and create a mathematical model which predicts some behaviors of the factors that influence lifespan. I also come to a conclusion which for me was really unexpected. The model only holds under the assumption that the resource investment into longevity per body size decreases the larger the animal is. This builds a bridge to ideas i had thought of earlier which i will go into detail in chapter 3.
"3. Properties of Investment in Longevity" maybe is the most interesting part. Here i talk about what properties different types of damage to the body have, how nature has already solved the quest for biological immortality and why it is so rare. I also paint a picture of how organisms try to extend their lifespans and present a mechanism by which the just mentioned assumption holds.
"4. Relation to Evolutionary Genetic Models of Aging" explains the leading evolutionary theories of aging and why there is something missing.
1. Adaptation: Nature’s Strategy for Survival and Success
1.1 Introduction
Life can be viewed as information with the function of self-preservation. Any informational pattern that causally influences its environment to favor its retention will, by default, be overrepresented in the future. In this light, living systems are essentially engines for the preservation and propagation of the informational units they embody.
In the evolutionary landscape, genes are those fundamental units whose survival depends on their capacity to propagate, not only within isolated individuals but also throughout an ever-changing gene pool of a species. From this gene-centered perspective, each individual organism serves as both a vessel and an environment where genes execute functions that ultimately enhance the host’s fitness. Natural selection, then, acts by promoting gene functions that improve an individual’s average likelihood of survival and reproduction, allowing those genes to persist and spread through the population.
Sexual reproduction plays a pivotal role in this gene-centric framework by facilitating the fusion and reassortment of informational units across distinct lineages. Through the processes of meiosis and fertilization, genes that have evolved in different organisms are brought together within a single zygote, effectively rendering other individuals into components of a gene’s selective environment. This genetic interchange generates novel combinations of traits, thereby increasing the raw material upon which natural selection can act. In this way, sex operates as both a mechanism of variation and a conduit by which genes expand their influence beyond the confines of any one host, weaving a dynamic network of interdependent individuals that shape the context of selections influence. In this landscape, genes do not merely adapt to environments, but they co-construct the way in which they get created and selected. The appearance of higher-level selection, such as that of groups or species, often emerges from this logic. Genes can engineer conditions that favor their own persistence, even when those conditions span multiple individuals and seem to benefit collectives. They can also appear to be destructive to themselves and other genes within the individual.
1.2 Adaptation in engineering
In engineering, the power of adaptation reveals itself most vividly through rapid prototyping: when designing a new engine or component, the fastest way to uncover hidden flaws is to build and test a working model rather than endlessly refining designs on paper. Mentally, we simulate and refine ideas against our internal world model, assessing their fitness in that limited environment. Yet the true proving ground is the laboratory. Only there, under genuine operational stresses, does a physical prototype reveal its unanticipated weaknesses.
This iterative cycle—conceive, build, test, learn, and refine—allows engineers to rapidly evolve their designs toward optimal performance given the requirements. By direct analogy, in biology ‘conceive’ maps to DNA variation and allele shuffling via sex; ‘build’ to organismal development; ‘test and learn’ to the adult’s trial in nature, where fitness is measured by survival and reproduction; and ‘refine’ to the inheritance of successful variants and the next round of variation beginning from a fitter foundation.
In industry today, rapid manufacturing techniques like 3D printing and CNC machining have accelerated this process, enabling companies to explore bold new solutions, respond nimbly to shifting market demands, and cultivate innovations that might otherwise lie dormant in the realm of pure theory. By embracing fast feedback loops the pace of innovation has increased in recent years.
1.3 Mechanisms of adaptation
When two co‐occurring species vie for the same ecological niche and with it, the same resources, under fluctuating environmental conditions, the lineage that can most rapidly generate and screen adaptive variants, and thereby unlock previously inaccessible resources, gains a decisive edge. This dynamic is captured by the extended Tangled Bank Hypothesis, which posits that sexual reproduction is advantageous because it produces genetically diverse offspring capable of exploiting ecological opportunities that are temporarily or spatially underutilized, allowing the population to more fully occupy the available niche space and outcompete less variable lineages.
In natural communities, competitors, predators, parasites, and even mutualists all become part of one another’s environment, and because these interacting species are themselves evolving, most lineages inhabit ever‐shifting adaptive landscapes. Within each species, this arms race creates a selective niche for evolvability‐modulating genes. The most famous ones, the mutator-alleles transiently elevate the genomic mutation rate, thereby accelerating the exploration of DNA-sequence space and hitchhiking to high frequency alongside beneficial mutations. Viewed at the population level, the gene pool thus functions as a collaborative experimental arena to continually seed new individuals carrying fresh mutational and recombinant prototypes into the ecological trial, amplifying those that perform well and culling those that do not. In this way, natural selection not only sculpts organismal phenotypes but also hones the very mechanisms of variation themselves, tuning the rate and nature of evolutionary change in response to shifting selective landscapes.
According to this narrative, one should find countless mechanisms that generate variation, others that make this process more efficient, and still others that detect when increased variation is required followed by ist upregulation.
Transposon activity is kept in check by multiple, overlapping silencing mechanisms like DNA methylation, transcriptional repressors, and small RNAs, but a controlled window of epigenetic derepression occurs at defined stages of wild-type germline development. Rather than a by-product of chromatin remodeling, this tightly regulated reactivation appears to be programmed. Environmental stress activates transposons in plants, heat shock activates P-element transposons, leading to increased mutation rates in Drosophila melanogaster and even pathogen attack can directly stimulate retrotransposition events in rice. Transposons, then, should not be seen merely as parasitic elements, but as part of a controlled mechanism for generating genetic variation when it is most likely to be beneficial.
In many organisms, stress‐sensing regulatory pathways trigger a boost in male production, thereby shifting populations toward outcrossing and greater genetic variation for faster adaptation. For example, most cyclical parthenogenetic animals, like Daphnia often start to reproduce sexually when environmental conditions deteriorate. In C. elegans, acute environmental stressors, such as heat shock, starvation, or irradiation directly elevate X-chromosome nondisjunction during hermaphrodite meiosis, leading to a rapid surge in XO male offspring in the very next generation. Experimental evolution under increased mutation rates or novel environmental challenges further selects for higher outcrossing, demonstrating that stressors drive up male production to boost genetic variation and facilitate rapid adaptation.
In a sexually reproducing population aiming to generate genetically diverse offspring better suited to fill the available niche space, the decision of which genes to combine in creating a new individual is critically important. Mechanisms to detect suitable genes should therefore be expected. For example, many species, including humans, show mating preferences influenced by MHC-linked odor cues, which can promote offspring heterozygosity and improve resistance to disease or reduce inbreeding risk. While MHC genes are a well-studied example, recent evidence suggests that genome-wide genetic variation can also shape individual odor profiles, indicating that many different genetic loci may contribute to scent-based assessment of genetic compatibility.
By embedding self-organizing, “follow-the-environment” programs into development, a single genetic tweak to bone length or shape can automatically propagate through multiple interacting tissues. This would greatly amplify phenotypic impact with minimal genomic change. For example, the developing skeleton instructs the directional outgrowth of skeletal muscle and other soft tissues during morphogenesis. In a similar way, blood and lymphatic vessels expand and branch into reshaped or growing tissues by responding to broadly conserved growth factors and extracellular matrix cues. Their architecture emerges from local conditions, not from explicit, tissue-specific genetic instructions. This allows vascular networks to dynamically adapt to shifting anatomical landscapes with minimal genomic reprogramming. Together, these generic self-assembly rules let organisms trial large morphological variants with only a small upstream mutation, confident that the surrounding connective, muscular, and vascular systems will “fill in” appropriately—accelerating the search through morphospace without needing countless downstream gene changes.
2. Model for Adaptive Gain
Building on the extended Tangled Bank framework, we now see that if the race between species comes down to who can most rapidly generate and screen new adaptive variants, then the total investment a species devotes to producing one adult must appear as a limiting factor in any function describing its rate of adaptive gain. This investment (Cₐ) entails not only the energetic and material costs of development, but also the time required to reach maturity, a crucial factor, as time spent developing is time not spent reproducing. Species with shorter generation times can iterate through adaptive cycles more quickly, gaining a potential advantage in the evolutionary race.
Alongside the cost of producing each adult, we must also account for the adaptive potential that every new individual brings into the race. Let’s call this quantity G, the expected evolutionary benefit yielded by one freshly matured organism. In a dynamic and ever-changing evolutionary landscape, the significance of G becomes increasingly pronounced. Each lineage is engaged in a continual struggle for survival, compelled to perpetually refine its genetic makeup in response to the adaptations of competing organisms. This ongoing cycle of mutual adjustment underscores the constant pressure to evolve simply to maintain relative fitness. Should G dip below this critical threshold, a species will inevitably be overtaken by its competitors and face extinction. In an environment teeming with small, readily accessible improvements, like minor tweaks to metabolic pathways or shifts in the dimensions of extremities, each offspring has a high probability of carrying a beneficial variant, so G becomes large. In contrast, when the fitness landscape is rugged, with isolated peaks separated by deep valleys, the situation changes. The population may sit atop one peak, but the likelihood that any single individual will discover a truly helpful trait, with which it reaches a higher peak, is low. In such cases, G remains small. Crucially, G depends not only on the raw genetic variation generated by mutation and recombination, but also on how that variation maps onto real‐world challenges: predator evasion, resource exploitation, or pathogen resistance.
In order for G to function as a proper term in our model, it must carry units that reflect the expected adaptive benefit per offspring per unit time. Concretely, we can think of G as the amount of “new resource‐access capacity” that one freshly matured individual on average with its genetic variance contributes, divided by time. A larger individual, having required greater investment to develop, must acquire more resources in order to compensate for its higher production cost. We now have in hand a expression for a lineage’s cumulative adaptive benefit over time:
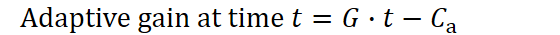
Here, G captures the potential per‑individual resource gain each adult is expected to contribute per unit time, while Cₐ represents the one‑time setup cost required to raise a newborn all the way to reproductive maturity. Every new adult effectively runs its own evolutionary experiment. The benefit term grows linearly with time, whereas the maturation cost is paid only once.
It is important to emphasize that the relationship formalized here holds under a specific ecological and evolutionary condition: a background of ongoing environmental and biotic change. In such a setting, stability is illusory: competitors, predators, parasites, and the environment itself are all in flux. Adaptation is not a one-time event but a perpetual process. The framework assumes that selective pressures never settle, so the need for continual innovation remains fixed. Only under these conditions, where evolutionary stasis is not an option, does the balance between the adaptive potential of new individuals (G) and the cost of producing them (Cₐ) determine a lineage’s capacity to keep pace and persist.
In a hypothetical world without any penalty for living longer, the most successful lineages would evolve individuals capable of indefinite reproduction. A single, immortal adult carrying an optimal allele combination could produce an ever-increasing number of descendants. This would reduce the possibility of meiosis and chromosomal crossover to break up this advantageous combination and with that minimizing the risk of adaptations being lost. By continuously producing offspring, that one genotype would fix itself in the population faster than any gene combination constrained by generational turnover. This genotype would then only be displaced if a new one arose whose offspring were even better suited to the current environment, gradually replacing the original through superior survival and persistence.
Yet in nature we basically never observe infinite lifespans, suggesting there must be a cost to extending longevity. To capture this, let CL denote the per‑unit‑time burden of staying alive and reproductively active. Each extra moment of life adds not only more opportunity to generate offspring (and thus more “gain”) but also this accumulating cost. Chapter 3. takes a closer look at what properties this cost has and what mechanisms stand behind it. Our formula therefore becomes:
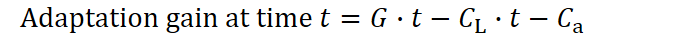
Longevity investment (CL) defines how much a species allocates toward extending an individual’s reproductive lifespan. A larger CL pushes the potential lifespan boundary farther out, up to, say,
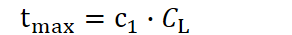
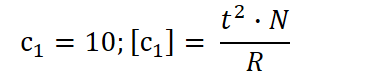
with N being the number of individuals within a species and R the resources. A high CL allows that individual to continue contributing offspring (and thus adaptive gain) for many more time units. The constant c₁ is not fixed; it can be shaped by both evolution and environmental context. Embedded within it are mechanisms and strategies that enhance the efficiency of longevity investment, like biological innovations that allow organisms to extract more value from each unit of CL. External factors may also modulate c₁. For instance, ambient temperature plays a significant role. In extremely cold environments, certain species, such as the Greenland shark, achieve remarkably extended lifespans, likely due to slowed metabolic rates and reduced cellular damage over time.
However, because the same resources devoted to longevity could instead have been poured into earlier reproduction, a high CL depresses early‐life adaptive yield: individuals invest more in maintenance, so they reproduce more slowly. Conversely, a low CL frees up resources for rapid, front‐loaded reproduction, producing a sharper early‐life gain, but it also means a much shorter horizon because the lifespan ceiling is low.
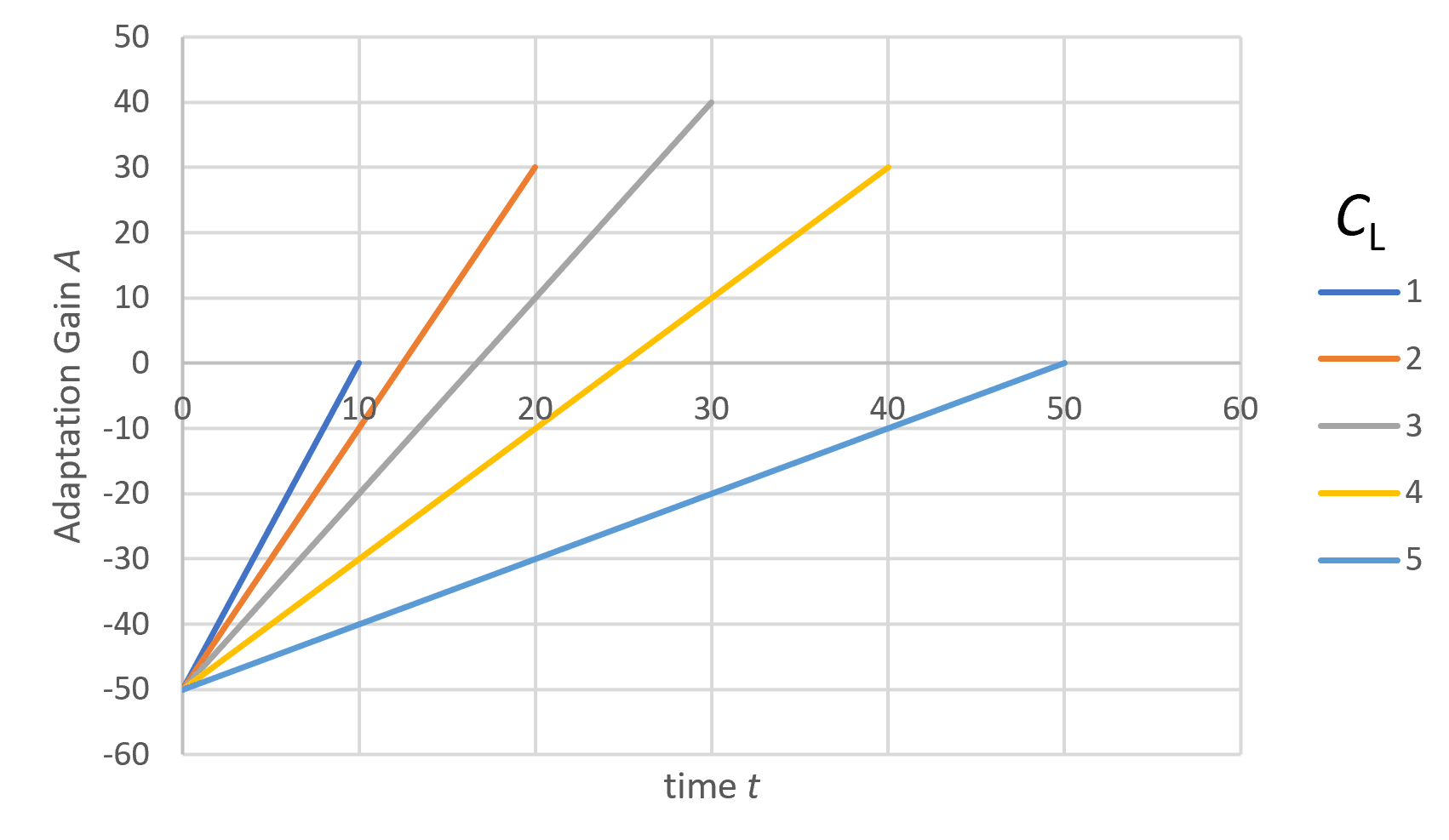
In figure 1, we see examples of different strategies by varying CL while keeping G = 6 and Cₐ = 50 constant. Among these strategies, the highest adaptation yield is achieved with CL = 3, corresponding to a maximum lifespan of 30.
To visualize only the final outcomes (endpoints) of each strategy, we evaluate the adaptation yield at the maximal lifespan defined by each CL, using tmax = c₁·CL, with c₁=10. This gives us the formula:
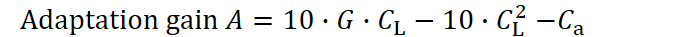
For a single generation, this function reaches its maximum at CL = 3. However, if the average achievable lifespan decreases due to external pressures such as predation (as shown by the red line in Figure 2), then a CL of 3 is no longer optimal. In such conditions, the species gains more by evolving a faster, shorter-lived strategy that delivers higher early-life returns. This pattern has been found in a lot of different animals. But it is also hard to distinguish to the influence of G, which when increased also reduces the optimal investment in longevity as shown later in this section. With a predator posing as an additional selection pressure G would increase.
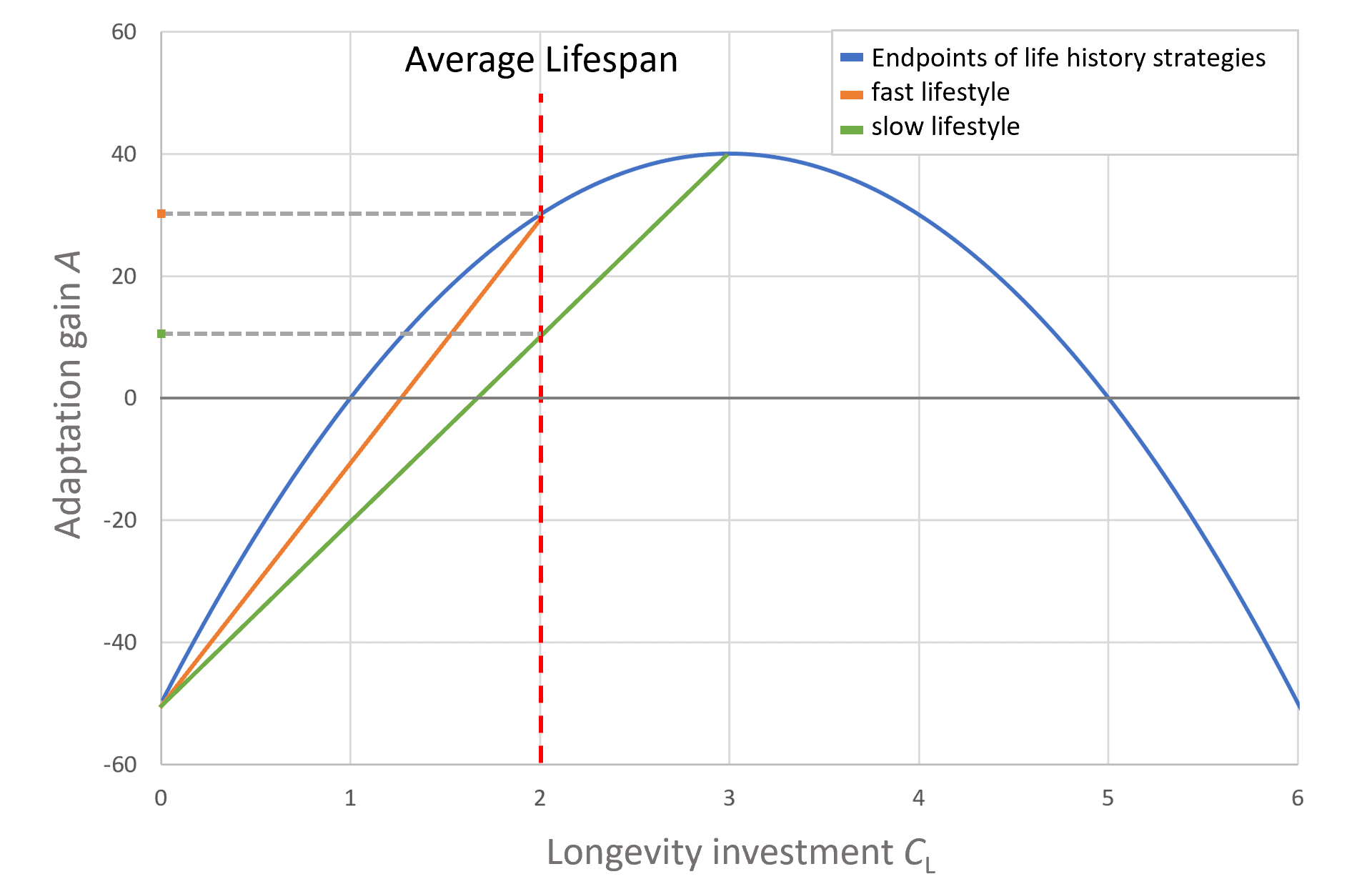
In this model, the adaptation yield remains constant over time starting from an initial investment in somatic development to reach maturity (Cₐ). However, many organisms adopt a strategy of continuous growth, enabling them to produce more offspring at later stages of life. In such cases, the yield term would be better represented by a power function, reflecting increasing reproductive output with age due to continued growth. Additionally, a separate term accounting for the energetic cost of continued growth would be required. These dynamics, however, are beyond the scope of the current analysis.
For a species to increase body size, greater investment in the component Cₐ is required. However, for this investment to be effective and for the adaptive gain to remain greater than zero, the adaptive potential G must increase alongside Cₐ. Consequently, a larger individual must be capable of converting a greater amount of environmental resources into its own biological structure, effectively reorganizing matter into more of itself. This ensures that the increased investment in body size results in a net adaptive benefit. This dynamic enables the attainment of significantly greater body size and lifespan. Figure 3 illustrates this relationship.
When considering only the costs associated with longevity (CL) and those associated with investment to create an adult (Cₐ), there exists an optimal lifespan for each value of Cₐ that minimizes total cost. The total cost can be expressed as:
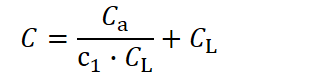
with c₁ as the constant representing the efficiency with which investment in longevity extends lifespan. The optimal investment in longevity that minimizes this cost function is given by:
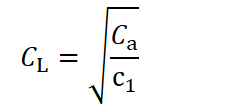
Thus, it follows that the larger an organism is, the longer it must live to pay back the resources invested in its development. However, one crucial aspect has so far been neglected: in our current formulation, the per‑unit investment in longevity is assumed to be independent of body size. In other words, if two organisms of different sizes share the same lifespan, the larger one would appear to require fewer resources per unit size to maintain that lifespan. Biologically, this would imply that an organism built from more cells becomes cheaper per cell to sustain for a given duration. To make this explicit, let us now allow the longevity‐investment term, CL, to scale with body‐size investment, Cₐ. One simple way to capture this is:
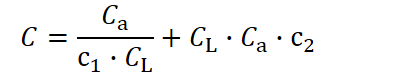
Where c2 is a constant that determines how strongly the cost of longevity correlates with the body‑size investment. Crucially, in this formulation the value of CL that minimizes the total cost,
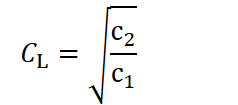
is independent of Cₐ. In plain terms, the optimal magnitude of longevity investment does not change with an organism’s size. Thus, if one seeks to explain the evolutionary possibility for species to evolve ever-increasing body sizes and with that a higher maximum lifespan, one must assume that investing in longevity is either independent of size or becomes more cost-effective as size increases. In chapter 3. the biological meaning of CL and mechanisms that could fulfill these conditions are explored.
In nature, reproduction early in life carries extra weight because offspring born sooner not only contribute immediately to population growth but also themselves begin reproducing at earlier ages, effectively compounding the lineage’s expansion over time. The intrinsic rate of natural increase (r), determined by solving the Euler–Lotka equation, is the most commonly used metric in life history theory.
By recasting the Euler–Lotka framework in terms of our adaptation‐focused payoff, we treat the classical term ℓ(a) b(a)—survival to age a multiplied by fecundity at a—as the net adaptive output of an individual:
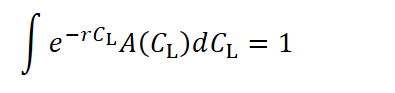
Figure 4 illustrates a representative example of how the optimal lifespan strategy shifts when successive generations are considered. The orange line shows where the optimum is, considering only a single generation.
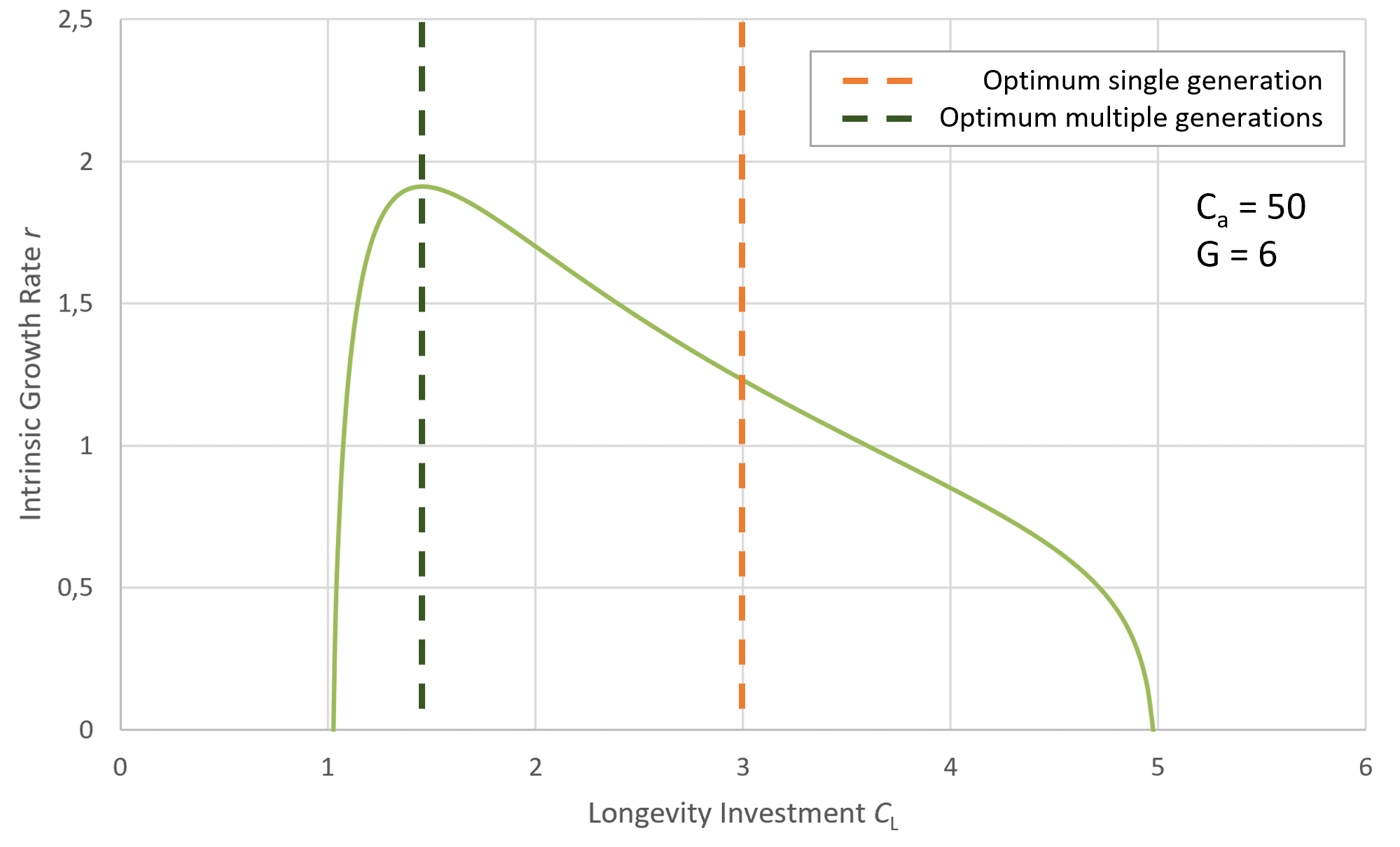
Figure 5 shows the effect of increasing developmental investment Cₐ while holding the adaptive potential G constant. As Cₐ grows, the cost of reaching maturity rises, pushing the lifespan optimum further toward later ages. Importantly, any lineage that can reduce Cₐ without incurring a fitness penalty will be driven to do so, since a lower Cₐ directly increases the intrinsic growth rate r.
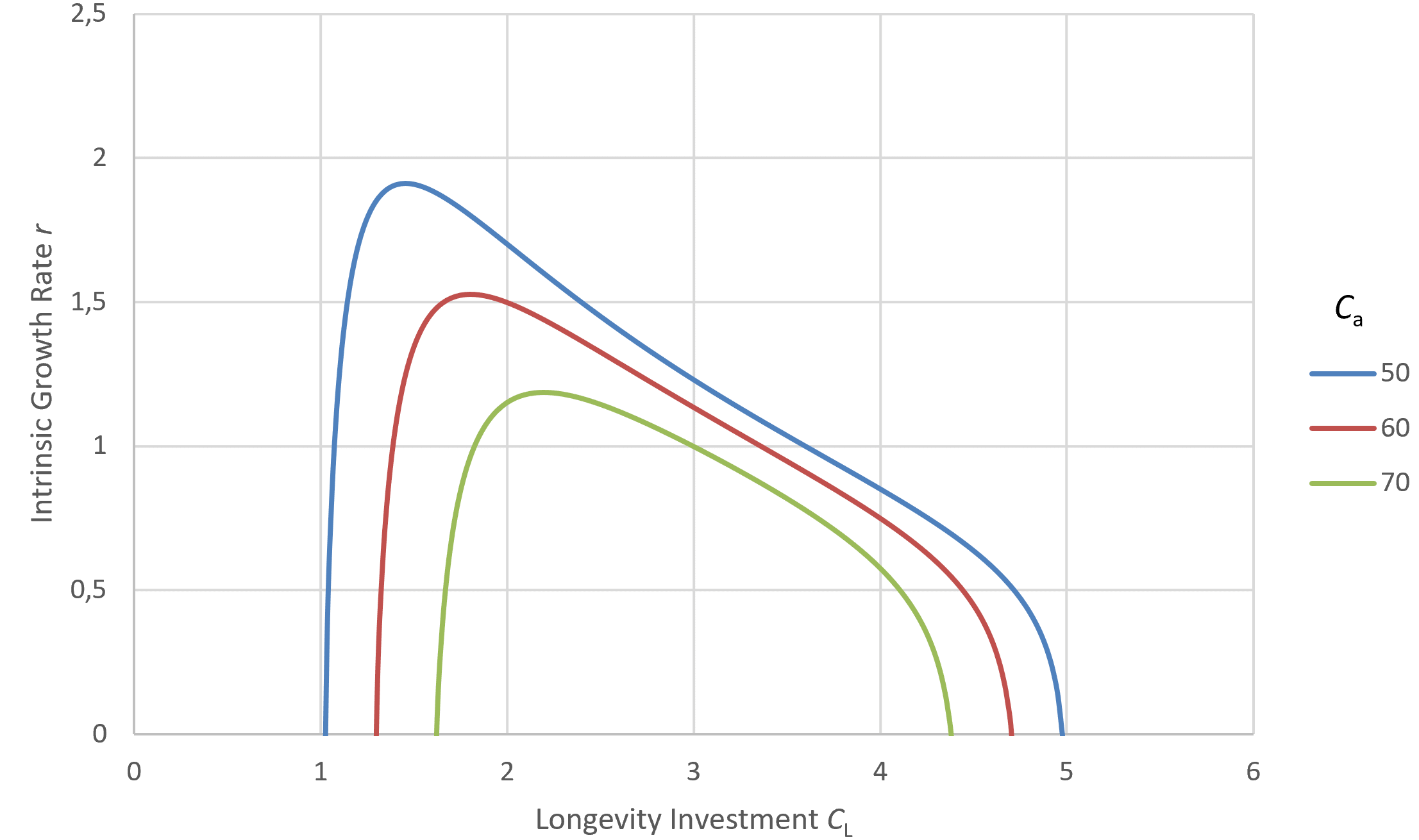
Figure 6 depicts the impact of varying G at fixed Cₐ. Changes in G, for example, from environmental shifts that free up new resources with minimal additional adaptations, makes shorter maximum lifespans more advantageous and thus favors the evolution of briefer life histories.
3. Properties of Investment in Longevity
3.1 Types of damage
Damage to somatic tissue can broadly be categorized into two groups. The first are external insults to an otherwise healthy body, for example ultraviolet radiation causing a sunburn, or a laceration or abrasion to the skin, or even a fractured bone. Whenever a particular type of injury recurs frequently within a species, such that it compromises an essential function, there is a clear evolutionary incentive for genes to evolve that restore that function starting from the damaged state. Over time, this pressure can give rise not only to repair mechanisms that patch up minor wounds but to astonishing regenerative feats: an octopus re‑growing an entire arm, or the human liver restoring itself after substantial tissue loss. At its heart, the process is one of cost versus benefit: only if the energetic and regulatory expense of maintaining and deploying an repair pathway is outweighed by the survival or reproductive gain will those genes be retained and refined across generations. When a specific type of injury never happens in nature, an organism will not evolve mechanisms to repair that injury.
By contrast, the second category of damage is internal and continuous. Every living cell faces a background of molecular insults, reactive oxygen species generated by normal metabolism that oxidize lipids and proteins, nascent polypeptides that occasionally misfold or aggregate, or metabolic by‑products that, if allowed to accumulate, would become toxic. To counteract this steady trickle of injury, organisms deploy a suite of constitutively active maintenance systems. There are molecular chaperones to shepherd proteins back to their proper shapes, proteasomes and autophagic pathways to clear irreparable debris and antioxidant enzymes to scavenge free radicals before they inflict further harm. Because these internal threats are unrelenting, any genetic variant that accelerates turnover of damaged molecules or enhances repair capacity yields a continuous fitness advantage, driving the evolution of comprehensive, energy‑balanced quality‑control networks that safeguard cellular integrity around the clock.
Both categories share a crucial feature that the lost function can be fully restored at sufficient energetic cost. A fractured bone will eventually knit back together, and active proteolytic systems can completely clear away protein aggregates. Aging, by contrast, is characterized by an system wide decline in nearly every physiological system, one that repair pathways cannot wholly reverse. Because the functional losses of senescence never return to their youthful baseline, the damage of aging cannot be neatly slotted into either of the two prior categories. Instead, it represents a unique, cumulative breakdown, in which repair mechanisms themselves gradually lose efficacy.
At first glance, this gradual decline evokes the familiar "wear and tear" of everyday objects and machinery like material fatigue, abrasion, corrosion and aging research often frames its focus in analogous terms. Proteins aggregate like debris in clogged filters, stem cell pools dwindle much like battery capacity and telomeres shorten as if the bristles on a brush frayed over time. These examples fit neatly into the wear‑and‑tear paradigm. Yet there is a crucial distinction: unlike inanimate parts, the body continually renews itself. Every cell, protein filament, and tissue type turn over at a characteristic rate far shorter than the organism’s lifespan.
The mammalian intestine presents a dramatic case, turning over as rapidly as in two to four days. Despite the remarkable longevity of most mammalian neurons, which can persist for over a century, their molecular components are in constant flux. While the neuron itself is largely irreplaceable, its constituent proteins continuously turn over with the large majority of proteins having a lifetime between 3 and 13 days.
Exceptions
There are some parts of the body with a really low turnover rate like elastin which was already mentioned here.
There does seem to be a turnover rate which is in humans close to their typical lifespan. I do not see any reasons why there could not just evolve proteins and mechanisms that would repair damaged elastin in the extracellular matrix when it would be needed and witht that give a fitness advantage. There also seems to be huge differences in elastin turnover in different organs in this example in hamsters.
Yet this perpetual renewal does not yield ever-fresh components operating at peak performance. One would expect that replacing old parts would restore flawless function, but during aging each renewal cycle simply substitutes a suboptimal element with another of equivalent deficiency. Over years of continuous turnover, these uniformly underperforming replacements accumulate, gradually degrading the system’s overall functions.
Aging thus represents a type of damage that, once lost, cannot be restored. In any self‑maintaining system, repair depends on components that carry within themselves the information required to rebuild damaged parts. The only elements that meet the criterion of irreversibility are those whose own blueprint is contained intrinsically. Once that information is destroyed, it cannot be recovered. In other words, if you obliterate a structure along with the self‑encoded instructions needed to recreate it, full restoration becomes fundamentally impossible. Across the tree of life we observe enormous variation in natural lifespans, which suggests that the pace of aging is itself evolvable. If aging truly reflects the gradual build‑up of irreparable damage, then some categories of age‑related injury must be either minimized or deferred in long‑lived species.
3.2 Loss of Information
One of the most fundamentally irrecoverable information stores in any multicellular organism is its genomic DNA. When somatic mutations disable critical genes, the collective performance of tissues and organs inevitably declines. Fortunately, evolution has equipped cells with a variety of safeguards like DNA‐repair enzymes, proofreading polymerases, chromatin‐remodeling factors, and damage‐sensing checkpoints, that slow the accumulation of these mutations. Every joule of metabolic energy devoted to nucleotide excision, base‐pair correction, or removal of cross‐links reduces the burden of DNA damage. By investing resources in these protective and maintenance pathways, organisms can prolong the integrity of their genetic blueprint and with it, sustain tissue function for a longer span of time.
Another key layer of information that has garnered intense interest in recent years is the epigenome. Like mutations in the DNA sequence, epigenetic alterations (epimutations), such as changes in DNA methylation, histone modifications, or chromatin organization, can impair cellular and tissue function when they stray from the cell-type-specific pattern.
The central importance of maintaining epigenetic integrity is underscored by multiple lines of evidence. For instance, in mammals, a lower rate of epigenetic drift correlates with a greater maximum lifespan. In model organisms such as C.elegans, fruit flies, and mice, aging is consistently associated with a loss of heterochromatin. These are regions of tightly packed, transcriptionally silent DNA. Loss of heterochromatin is accompanied by global transcriptional noise. If a fully differentiated neuron, for example, inappropriately expresses genes meant for a stem cell or a muscle cell, it not only wastes energy but also undermines its specialized role. If the function of a particular cell type or tissue declines beyond a critical threshold, other organs or systems that depend on it may also begin to fail, potentially leading to systemic collapse and, ultimately, death.
Remarkably, somatic cells from aged individuals, when used for cloning, can give rise to entirely new organisms that develop normally and live full, healthy lifespans.
Collectively, these findings are consistent with a central premise of the epigenetic information theory of aging (ITOA): that the progressive loss of epigenetic structure contributes to the erosion of cellular identity and function. However, other aspects of the theory remain speculative and are not adopted here. For example, ITOA proposes that cells retain a complete, intact backup of their youthful epigenetic information, which can be restored through partial reprogramming. This notion raises several conceptual and biological concerns. It is unclear how such a backup system could be maintained over time without being subject to the same environmental and molecular insults that drive aging in the first place. Additionally, the hypothesis that activation of endogenous Yamanaka factors could rejuvenate the organism overlooks a key evolutionary question: if such mechanisms exist naturally, why have they not been selectively utilized to preserve function and extend lifespan? This would fall into the other two previously named category of somatic damage. While whole or partial reprogramming can clear epigenetic marks in experimental contexts, this does not necessarily imply the presence of an innate, organism-wide rejuvenation program.
In principle, the blueprint for the correct epigenetic state resides in the genome itself, yet mature cells lack direct access to the developmental programs that originally established those patterns. During embryogenesis and early differentiation, cascades of developmental transcription factors and signaling pathways choreograph a highly complex program that both specifies cell identity and locks in its epigenetic marks. In this process, cells rely on intercellular communication, with neighboring cells sending instructive signals at precise developmental time points to guide lineage decisions and establish the appropriate epigenetic landscape (This video is a great example to get a feeling for the complexity of development). Once this program has run its course and cells settle into their final spatial locations, they cannot simply replay it to reset their epigenome.
Because the original developmental machinery cannot be reactivated in adulthood, any epigenetic drift that occurs afterward is effectively irreversible, much like a mutation in the DNA sequence. To counteract this gradual loss of information, organisms must evolve dedicated epigenetic maintenance systems, including genes that encode chromatin‑binding proteins, modification enzymes, and structural factors that preserve cell‑type‑specific patterns. As with DNA repair, such protective mechanisms can only evolve and persist if the benefits of preserving cellular identity exceed the energetic costs. The loss of epigenetic information, therefore, mirrors genomic damage in a fundamental way. It cannot be fully reversed once the original state is lost, only slowed down by continuous maintenance.
3.3 Rejuvenation requires re-execution of developmental programs
A fully “young” epigenetic landscape in a differentiated cell can only be restored by re-running, in whole, the organism’s developmental program or by an equivalent process in which cells are first reset to an fully undifferentiated state and then allowed to proceed autonomously toward full differentiation. In effect, one must recapitulate the cascade of transcription factors, chromatin remodelers, and signaling events that originally established lineage-specific epigenetic patterns during embryogenesis. Without such a reset, mature cells have no direct means of erasing accumulated epigenetic drift or repositioning their chromatin to a truly naive state.
One of the most striking examples of such a reset in the animal kingdom is the so-called immortal jellyfish, Turritopsis dohrnii. When faced with stress, injury, or senescence, this hydrozoan can revert its mature medusa cells into a cyst-like structure, essentially a colony of pluripotent cells. From this cyst, it can re-differentiate into a new polyp colony, thus jumping back to an earlier point in its life cycle. As long as no new deleterious genetic mutations arise during its lifetime, the process can, in principle, occur indefinitely, allowing the jellyfish to escape the normal limits of somatic aging.
A parallel phenomenon occurs in many higher plants through vegetative propagation. At the tip of every shoot lies a meristem, which is a perpetual fountain of undifferentiated cells programmed to generate new leaves, stems, and roots. When a cutting is taken from this young tissue, both the epigenetic “age” of the cutting and the roots it subsequently produces are effectively reset to zero. Repeatedly planting new cuttings can extend a single genetic individual far beyond the lifespan typical in nature.
Some clonal species, like the quaking-aspen colony known as Pando, are capable of surviving for thousands of years by continually renewing themselves through the natural formation of genetically identical ramets.
In contrast to most vertebrates, certain invertebrates, most famously planarian flatworms, possess a regeneration program that functionally mirrors embryonic development. When a planarian is bisected, each fragment activates its resident totipotent neoblasts to rebuild all missing cell types and structures, restoring the organism to its original size and form.
This reconstruction proceeds through waves of proliferation, migration, and differentiation that are similar to early development, thereby endowing every newly formed tissue with a fully “young” epigenetic landscape. Because no part of the epigenome is simply repaired in place, but instead, it is entirely rewritten as cells re-differentiate, planarian regeneration effectively resets its age.
Other worm-like species, such as certain nemerteans and annelids, use analogous stem‐cell–driven cascades to achieve comparable feats of whole‐body renewal, demonstrating that a developmentally equivalent program can permanently forestall epigenetic drift so long as the underlying genetic instructions remain intact. The ability to regenerate both anterior and posterior body parts following dissection is often associated with an organism's inherent capacity for fission, a common mode of asexual reproduction in these animals. They therefore possess a genetic program that directs the formation of a new individual through a non-embryonic developmental pathway.
An important point to mention here is that with these species there is a functional decline visible over the timespan prior to fission. Even for the “immortal” freshwater planarian Schmidtea mediterranea shows alterations in sensory organs, loss of neurons and muscle, loss of fertility and impaired motility with age. But no overall decline of function is visible over successive fissions or after regeneration from injury because it leads to global tissue rejuvenation.
The only exception is if one looks at a successive line of heads. For example in Aeolosoma viride the head part has a definitive lifespan with a number of ~57 offspring produced via fission. In Schmidtea mediterranea lines of successive heads showed aging and ultimately died out. Conversely, in lines of successive tails, no aging was observed based on uniform fecundity and no recorded deaths. With each fission functionality is fully regained but not for all parts. Mechanisms by which this behavior could be achieved will be explored in the next chapter.
Biological immortality via a mechanism that resets an organism back to an earlier developmental state is extraordinarily rare and when it does occur, it only appears in very small, simple animals. Evolving such a mechanism requires not just small steps evolving over many generation which each give a fitness benefit that can normally be seen in evolution, but you would need the whole program to evolve before it gives any benefit at all and could be selected for. This is why no such mechanisms exist in large and complex animals because it is increasingly more unlikely that whole programs evolve for how to turn a fully differentiated body back into a undifferentiated state form where it could build a fully functional body again.
Negligible senescence in large animals
Negligible senescence is a term to denote organisms that do not exhibit evidence of biological aging (senescence), such as measurable reductions in their reproductive capability, measurable functional decline, or rising death rates with age.
Some fish, such as some varieties of sturgeon and rougheye rockfish, and some tortoises and turtles[4] are thought to be negligibly senescent, although recent research on turtles has uncovered evidence of senescence in the wild.[2]
In 2018, naked mole-rats were identified as the first mammal to defy the Gompertz–Makeham law of mortality, and achieve negligible senescence. It has been speculated, however, that this may be simply a "time-stretching" effect primarily due to their very slow (and cold-blooded and hypoxic) metabolism.[6][7][8]
For animals that keep growing it seems logical to evolve exceptional high lifespans because the reward when you make it to a high age pays off with lots of offspring. I do not have a good explanation for why in some animals no functional decline is visible with age. Because this observation does not fit with the loss of epigenetic information. I guess either senescence is stretched to a point where no measurements have been taken yet or there is some fitness gain over time like learning to survive better that counteracts the fitness loss .
In principle, then, the key to epigenetic “youth” lies in the capacity to re-execute a developmental program or its functional equivalent, allowing the organism to circumvent the otherwise irreversible drift of its epigenetic landscape. Accordingly, in sexually reproducing animals the germline periodically induces a state that permits a complete epigenetic reset and thereby rerunning its developmental program to return to its original, youthful condition. With that the germline effectively escapes cumulative epigenetic drift, loss of function and thus functions as an immortal cell lineage billions of years old.
3.4 Epigenetic repair mechanisms
There are two broad strategies by which a cell could safeguard its epigenetic information against continual assault. It is important to mention here that those strategies are completely different from programs to build new cells with a youthful epigenetic state like explained in the previous chapter.
The first is a biochemical stabilization approach. By favoring especially resilient molecular substrates the overall rate of damage and hence of epimutations is kept to a minimum. For example, a lineage might evolve histone variants that are less prone to oxidative modification. This adaptation would effectively fortify the epigenetic code itself, reducing the likelihood that stray chemical insults will permanently corrupt it. To minimize the assault itself, whether by shielding the information from sources of damage, lowering the concentration of chemical insults, or reducing metabolic activity, also falls into this category. For instance, lower temperatures can slow metabolic rates, thereby extending longevity.
The second is an information‑theoretic or software solution, in which redundancy and active maintenance routines continually rewrite and correct the epigenetic record regardless of the underlying damage rate. A straightforward example of this model involves transcriptional regulators that bind to their own enhancers or promoters, thereby reinforcing chromatin states that sustain their own expression. MyoD, a master regulator of the skeletal myogenic program in vertebrates, exemplifies this mechanism. It binds not only to the promoters of many muscle-specific genes but also to its own promoter, effectively locking the cell into a differentiated state. This self-reinforcing loop mirrors the behavior described by Waddington’s epigenetic landscape model where the cells try to stay in their own valley of differentiation.
Similarly, stable cell states can also emerge from small networks of transcription factors that not only self-activate but also mutually reinforce each other’s expression, creating a resilient system that can recover from epigenetic perturbations, as demonstrated in tri-stable regulatory circuits modeling T-helper cell differentiation.
On a smaller scale, molecular marks can also reinforce one another. H3K9me3, which recruits HP1, which in turn brings in SUV39H1 to add more H3K9me3 creates a self-sustaining silencing loop at specific genomic regions.
Epigenetic marks or transcription regulators that store and reinforce information collectively, can be thought of as multiple redundant copies, so that if one is lost due to an epimutation, the others can restore it. Multiple copies of essential epigenetic information also exist at a larger scale, specifically, at the level of cells. When cells of the same type communicate with one another, they may reinforce their epigenetic states, effectively linking their individual epigenomes into a coordinated network. Paracrine signaling and especially extracellular vesicles (EVs) can be a good mediator for this information flow.
EVs program the epigenomic profile by remodeling DNA, RNA, and histone modifications in recipient cells. This is achieved by delivering cargoes mainly targeting methyltransferase and demethyltransferase. EVs and epigenomic profiles are closely linked and reciprocally regulated. Even DNA has been found in Exosomes and it was shown that it can integrate into the genomes of recipient cells.
If one essential gene gets silenced by accident in a cell and all cells continually send information about what genes they express, then they can rescue the right gene expression in that cell again. In combination with the other self reinforcing strategies to retain its epigenome this intercellular communication helps maintain stability and identity across a population of differentiated cells.
To capture these ideas in a simple model, imagine each cell as a 5×5 grid of on/off switches, where each switch represents one gene: on (1) means the gene is active, off (0) means it is silenced. A perfectly differentiated cell corresponds to a grid in which exactly one entire row of genes is switched on and all others are off. With that there are 5 different cell types (Figure 7). Random epimutations cause individual switches to flip on or off, and without repair the cell will gradually lose that perfect row, leading to partial or full dedifferentiation. Within this model the cells are dependent on the other cell types executing their functions only in that spatial location where they are sitting. If too many of wrong genes are expressed or too many right genes silenced the system dies.
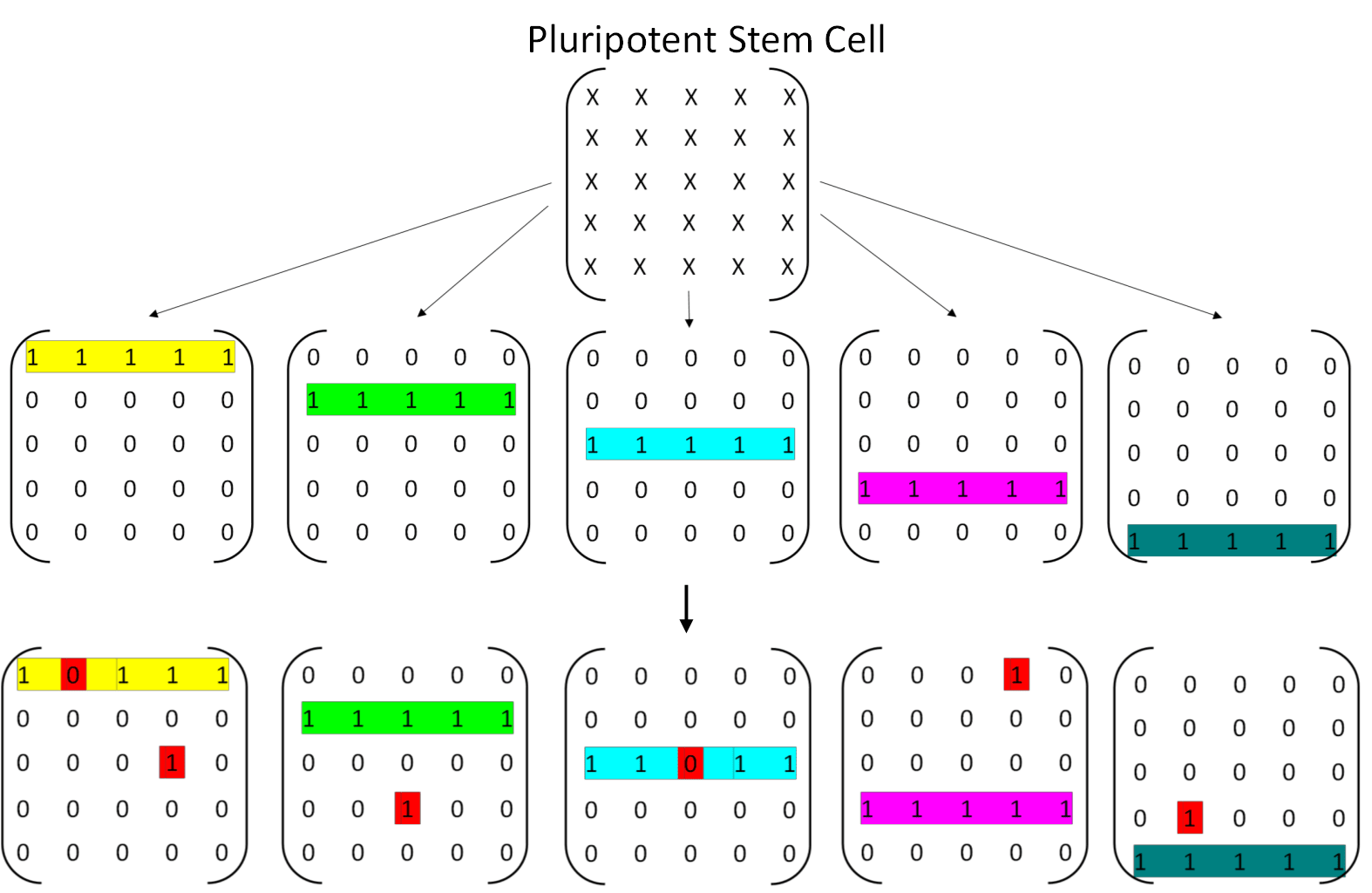
To conceptually capture the dynamics underlying Waddington’s epigenetic landscape, I propose a model in which each cell maintains a self-repair signal that depends sigmoidally on the number of active genes within a specific, cell type defining gene set, represented here as a row in a matrix. When most genes in this row are active, the self-reinforcement signal is strong, increasing the likelihood that any temporarily silenced genes are reactivated. As the number of active genes declines, the strength of this signal diminishes steeply, especially below a critical threshold.
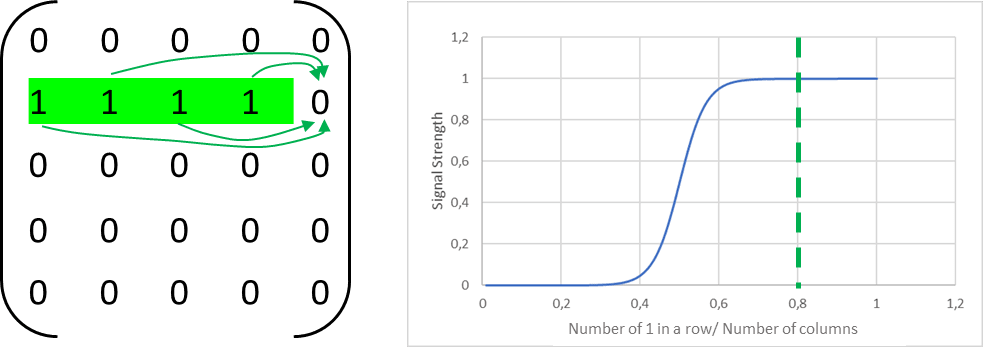
In isolation, a single cell subject to random epimutations will lose the ability to maintain this repair signal. Once enough genes in the active row are lost, the signal drops below a functional level, leading to the cell's dedifferentiation. This process has a measurable average lifespan. It is the time it takes for the cell to cross the tipping point beyond which it can no longer maintain its epigenetic identity.
The fact that epigenetic age can be measured based on specific, predictable changes at defined CpG sites, rather than a random erosion of epigenetic information, suggests that aging involves regulated mechanisms that shape the epigenome over time. These mechanisms likely act with varying strength across different genomic regions, leading to site-specific vulnerabilities or biases that, collectively, produce a reproducible pattern of epigenetic drift that can be quantitatively tracked. With this epigenetic clocks have been invented.
Now consider a pair of cells of the same type that are coupled via communication, such as through paracrine factors or extracellular vesicles (Figure 9). In this coupled system, each cell receives its repair signal not solely from its own gene activity, but also incorporates input from its neighbor’s state, weighted by a coupling strength. For instance, if Cell A’s gene set becomes degraded and drops to only two active genes, its self-signal would be weak. However, if Cell B still maintains near-complete activity, its strong signal can compensate, enabling Cell A to recover its gene expression profile. The same applies in reverse. The probability that both cells simultaneously lose their identity becomes substantially lower than the risk of individual failure, thus extending the overall duration of stable differentiation.
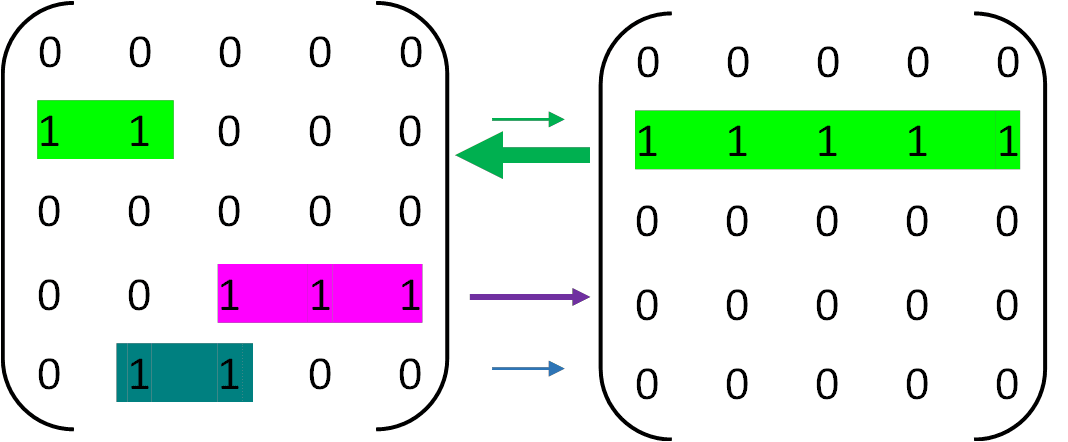
Importantly, cells do not inherently “know” which genes define their correct expression profile. They maintain the configuration given to them during development and propagate it through intracellular and intercellular signaling. If one cell's profile becomes corrupted, it still broadcasts this faulty state to others. Over time, this leads to a gradual decline in the average signal shared across the population, and eventually, the system’s capacity for self-repair collapses. Once a critical number of cells lose their specialized identity, the tissue or organism may no longer function properly—ultimately resulting in organismal failure and death.
Partial reprogramming
Partial reprogramming of cells with the use of the yamanaka factors has garnered a lot of attention recently. From here:
Three of four Yamanaka factors, including OSK, can restore
the transcriptome to a more youthful pattern. Organs that have
now been rejuvenated by OSK(M) reprogramming include kidney,
liver, skin, heart, brain and pancreas and muscle.
This is difficult to explain with this model. With expressing all the yamanaka factors cells will eventually lose their identity and turn back to a stem cell. This means the epigenetic information is completely lost. Doing a partial reprogramming would mean only a little information is lost. But this would still not be a good thing because aging would in theory still be accelerated. Even though partial reprogramming could so far not rejuvenate a whole organism, it still extended the lifespan of animals in some cases.
When i was programming and simulating this whole connection between the different cells i came to the following conclusion: With the assumption that there is a base rate of flips from 1->0 and 0->1 and if you wait long enough then there will be lets say a 50/50 ratio of 1 and 0. This is not something that you would want because the signal strength of one row could easily tip to very high and thus jump into another differentiation type. When you now program a function to increase the rate of 1->0 one gets to lets say a 10/90 ratio of 1 and 0, thereby minimizing the probability of one row accidently becoming full. The correct full row would also be degraded faster which could be prevented by increasing the frequency of self repair resulting from a nearly full row. Active (random) removals of epigenetic marks could thus be used to slow down aging.
Another possibility is that whatever is being measured in all those cases is not really aging. Aging is hard to measure and there is no consensus on what is the best method. Therefore you get questionable results like this: When a person has stopped smoking, their biological age (based on aging biomarkers) is decreased.
I think partial reprogramming can not be the whole answer.
Within this model, two straightforward strategies can extend the time before such collapse:
The first strategy is by increasing the number of genes in the critical row. By expanding the number of actively maintained genes, the system becomes more resilient to random mutations. More events are required for the repair signal to weaken. However, this strategy comes with increased metabolic and genomic costs (CL) , as more genes must be expressed.
One can expect that the activity of these maintenance mechanisms is, at least in part, subject to regulation. For example, they might be downregulated to conserve energy for reproduction during favorable conditions. Conversely, in times of environmental stress or resource scarcity, when the chances of successful reproduction are low, organisms may upregulate these mechanisms to invest in somatic maintenance and longevity, effectively preparing for a more favorable future opportunity to reproduce. This has been shown extensively by caloric restrictions and other stress factors extending the lifespan of organisms and a reduced fecundity. Importantly with this strategy aging can only be slowed down and not halted indefinitely like proposed in the disposable soma theory of aging.
The second strategy is by increasing the number of cells contributing to a shared signal. By expanding the number of synchronized cells of a given type, the system gains redundancy. When more copies of information synchronize with each other the way described in the model the time before all the cells lose their differentiation increases even though the amount of error brought into the system add up. Although this increases the initial investment required to build a larger population of cells (CA), the cost per cell to reach a specific age (CL/CA) decreases. This offers the previously mentioned explanation for how biological systems can achieve higher longevity with the maintenance investment becoming size-independent or more cost-effective as size increases.
when you are tall, you live shorter
At first glance, it seems logical to assume that being taller and therefore having more cells and more copies of the crutial information could lead to a longer life. This pattern generally holds true when comparing average lifespans across different species. The larger animals tend to live longer than smaller ones. However, the opposite trend often appears within a single species where larger individuals often have shorter lifespans.
Comparisons of different breeds of domestic dogs or individual dogs differing in size consistently show a negative correlation between adult body weight and longevity [12]. Very small dogs typically live over 15 years, while dogs from the largest breeds are not likely to reach the age of 10 years [10, 18]. Similar negative associations of body size and lifespan have been described in laboratory rats [19] and in domesticated horses [20], as well as in various human populations [21].
Those are domesticated and self domesticated animals. This seems important to me. That by explicitly selecting for size, other carefully evolved mechanisms could be crippled, resulting in a reduced lifespan.
There are also a lot of different factors influencing body size. For example mild caloric restriction can be followed by being smaller and living longer.
Beyond interactions within a single cell type, communication between different cell types may also serve a regulatory function in maintaining epigenetic integrity across tissues. Differentiated cells of one lineage can influence the epigenetic landscape of another, effectively adding up the connected cell types even though they are of a different kind. This forms a body-wide, interconnected signaling network through which information about cellular identity is continuously exchanged. It would not only be reinforcing local identity, but also preserving global tissue organization.
If aging is understood as the progressive loss of epigenetic information and consequently, of cellular identity then the content of these intercellular signals should likewise reflect an organism’s age. In this view, aged cells transmit “old” signals that may propagate destabilized or degraded identity patterns, while youthful cells emit “young” signals that reinforce proper gene expression states. Replacing aged signals with youthful ones would therefore be expected to restore, at least partially, the correct epigenetic configuration in older cells.
This conceptual framework is supported by findings from heterochronic parabiosis experiments, in which the circulatory systems of a young and an old animal are surgically joined. These studies show that young animals exposed to aged blood exhibit signs of accelerated aging, while older animals exposed to youthful circulation display molecular and functional rejuvenation.
In essence, the systemic exchange of epigenetic signals which are carried, for example, by extracellular vesicles or tissue-specific signals, can impose either a degenerative or regenerative influence, depending on the quality of the signal. This suggests that aging is not purely a cell-autonomous process, but one shaped by the collective informational environment of the organism.
While many studies refer to “youthful factors” as the agents of rejuvenation in heterochronic settings, such terminology often remains mechanistically vague. In contrast, the framework proposed here offers a precise rationale. Rejuvenation occurs not through a generic effect, but through the restoration of correct gene expression patterns. It is an informational reset driven by the reintroduction of accurate epigenetic signals. Rather than invoking undefined “youthful” substances, this model points to the return of coherent, cell type specific identity signals as the primary driver of systemic recovery. It is this informational fidelity and not simply molecular abundance of any kind that distinguishes young from old.
This perspective also helps explain why the previously mentioned freshwater planarian Schmidtea mediterranea regains full functionality following regeneration. The newly regenerated body parts are formed through a recapitulation of developmental programs, which inherently establish precise, youthful epigenetic marks. These newly patterned cells do not function in isolation; rather, they engage in intercellular communication with pre-existing tissues, transmitting epigenetic signals throughout the organism. In effect, youthful information is not confined to the site of regeneration but diffuses systemically, progressively rejuvenating the entire organism through coordinated signaling and identity reinforcement. The gradual loss of correct epigenetic information is overshadowed by the inflow of information from the newly built cells.
However, the flow of rejuvenating information may not necessarily be bidirectional—extending from the regenerated posterior to the anterior end. Genes would be needed for each direction the information flows to. This asymmetry could account for the observation that successive lines of head transplants in Schmidtea mediterranea exhibit signs of aging and ultimately died out.
It is conceivable that the quantity of correct epigenetic information originating from the newly formed posterior is insufficient to effect meaningful rejuvenation in the head region and is therefore overshadowed by epigenetic drift. Alternatively, certain specialized cell types within the head may remain functionally uncoupled from the posterior systemic signaling network, rendering them inaccessible to the youthful cues emanating from the tail and thus excluded from the regenerative rejuvenation process.
With this framework in mind, it becomes conceptually plausible that a single differentiated cell could be maintained in a functional and youthful state indefinitely, provided two conditions are met: first, that the intercellular signals reinforcing its identity remain youthful and coherent and second, that the underlying genetic material remains intact and free of deleterious mutations. This hypothesis could be tested experimentally by serially transplanting a cell or small part of an organ from an aged organism into a clonal youthful environment across multiple generations, thereby continually exposing it to a rejuvenating signaling milieu. These somatic cells could then have a much longer lifespan than the normal organism.
Extending this principle from a single cell to an entire organism suggests that sustained rejuvenation may be achievable by persistently replacing the signals involved in cell-to-cell communication. So long as the correct epigenetic patterns are reimprinted and stabilized within each cell. Generic, body-wide molecular or genetic interventions are insufficient, as proper rejuvenation demands the spatially targeted delivery of epigenetic information, thereby ensuring that, for instance, liver-specific gene programs are reinstated exclusively within liver tissue. If all cell types are interconnected, it may not be necessary to replace all signals because youthful information introduced into the system could ripple throughout the body and rejuvenate it, provided its renewal rate exceeds the loss from epigenetic drift.
4. Relation to Evolutionary Genetic Models of Aging
The mutation‑accumulation theory of aging proposes that senescence arises because natural selection grows ever weaker against deleterious mutations whose effects manifest only at advanced ages. Once an individual has survived past its prime reproductive period, the “selection shadow” cast by cumulative mortality renders late‐acting mutations effectively neutral, allowing them to accumulate in the genome unchecked. However, a major unresolved challenge for this theory is the absence of any clearly defined mechanism that times the activation of these harmful alleles. As Kirkwood (1977) has pointed out, without a credible molecular “clock” to delay expression until late life, the theory remains incomplete and circular, because it must assume just the kind of timing mechanism it tries to explain.
For any model in which gene activity depends on an organism’s age, there must exist some change or signal that reliably marks the transition from one life stage to the next. Antagonistic pleiotropy likewise rests on the premise that genes can switch their effects between “early‐life” and “late‐life” states, yielding benefits at one stage and costs at another. Yet this theory does not address why evolution could not favor alleles that simply mirror the beneficial early‐life signal indefinitely—thereby harvesting only the positive effects and wholly avoiding senescent decline.
A more nuanced perspective emerges once we consider the gradual loss of epigenetic information and the functional impairments that follow. Imagine a pleiotropic gene whose product boosts fitness. It is encoding a protein that makes a process more energy efficient but which naturally degrades over time, requiring cellular clearance mechanisms to remove its breakdown products. Early in life, efficient “cleanup” ensures continued benefit; later, however, as epigenetic drift degrades the very pathways responsible for debris removal, accumulating waste overwhelms the system and generates pathology. Because this failure occurs late and the positive effect persists throughout life, the gene is favoured by natural selection. It is also possible for a gene to accelerate aging but when it also gives a fitness gain that outweighs the negative effects it will fix itself in the population. A knockout of that gene would result in a longer lifespan.
The same logic applies to mutation‑accumulation: a mutation that increases the potential of the byproducts of the protein to aggregate at a certain concentration might escape purifying selection simply because its harmful effects manifest after most reproduction has already taken place. In both cases—antagonistic pleiotropy and mutation accumulation—the mechanisms they describe are secondary layers atop the fundamental processes that distinguish early life from late life. They explain how aging accelerates once the shift occurs, but the root cause remains the underlying transition in molecular and cellular integrity over the organism’s lifespan.
It is therefore critical to define and distinguish these layers with precision: the secondary effects described arise from an initial loss of function which is caused by the loss of information. Aging research should focus primarily on the root causes of functional decline, rather than uncovering the exact mechanisms of the functions that are ultimately lost or misregulated.
Discuss
