
2025年6月28日,来自上海科技大学等机构的白芳等在npj Drug Discovery上发表题为“Recent advances in molecular representation methods and their applications in scaffold hopping”的综述文章。

该文系统梳理了人工智能(AI)驱动的分子表征方法从传统规则到数据驱动的演进历程,重点分析了语言模型、图神经网络(GNNs)、高维特征、多模态学习和对比学习等前沿技术的最新突破,并深入探讨了这些方法如何革新骨架跃迁这一药物发现的核心策略,旨在加速新化学实体的发现。
药物发现是一项耗时且成本高昂的工作,研究人员不断开发新的实验和计算方法以加速药物研发的各个阶段。近年来,人工智能的进步使AI辅助药物设计成为研究热点,涌现出化合物成药性评估、虚拟筛选发现活性化合物、分子生成创建新化合物等前沿方法,这些方法在药物开发早期阶段发挥关键作用,能实现更快的早期筛选和潜在先导化合物的识别。
而这些方法的关键前提是将分子转化为计算机可读取的格式,即分子表征,它是机器学习(ML)和深度学习(DL)模型训练的基础,也是计算化学和药物设计的基石,架起了化学结构与其生物、化学或物理性质之间的桥梁。有效的分子表征对虚拟筛选、活性预测、骨架跃迁等药物发现任务至关重要,能实现化学空间的高效精确导航。
分子表征:从传统规则到AI驱动的创新
分子表征的定义与重要性
分子表征是将分子转化为算法可处理的数学或计算格式,以建模、分析和预测分子行为的过程。它不仅要编码化学结构,还需支持化学空间的高效探索。随着化学信息学和AI的发展,分子表征方法不断涌现,从传统的基于规则的特征提取,到现代AI驱动的学习连续高维特征嵌入,其演变极大推动了药物发现进程。
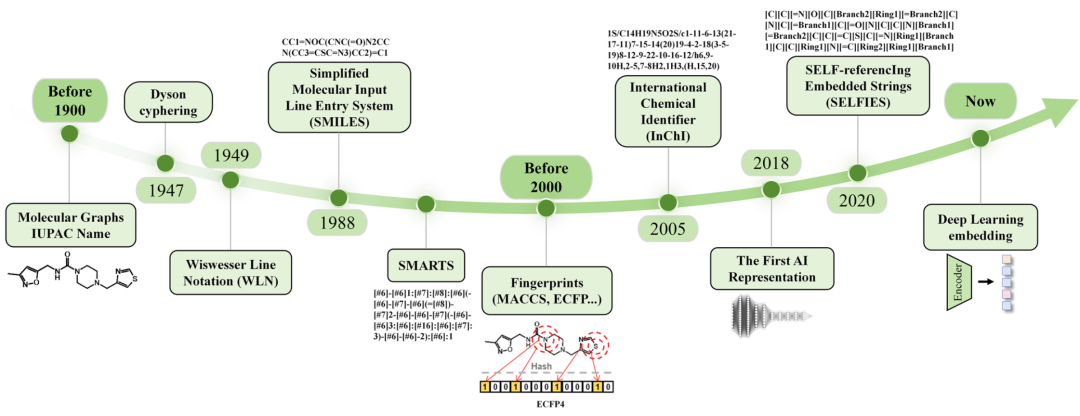
图1 各种分子表示方法的初步建议时间表
传统分子表征方法
传统分子表征方法依赖明确的、基于规则的特征提取,主要包括:
分子描述符:量化分子的物理或化学性质;
分子指纹:通常将亚结构信息编码为二进制字符串或数值,如广泛使用的扩展连接指纹,能紧凑高效地表示局部原子环境;
字符串格式:如简化分子线性输入规范(SMILES),1988年由Weininger等人提出,是一种紧凑高效的化学结构编码方式,后续的CXSMILES、OpenSMILES等均为其扩展。此外,还有IUPAC命名、Wiswesser线符号(WLN)等。
这些传统方法在相似性搜索、聚类、定量构效关系(QSAR)建模等任务中因计算高效和格式简洁而有效。但传统方法存在局限:SMILES难以捕捉分子相互作用的全部复杂性,且传统方法依赖预定义规则,难以反映分子结构与生物活性、理化性质等关键药物相关特征的复杂关系,在探索广阔化学空间时能力有限。
现代AI驱动的分子表征方法
随着AI的发展,基于DL的分子表征方法兴起,它们从大型复杂数据集中直接学习连续的高维特征嵌入,突破预定义规则限制,能捕捉分子的局部和全局特征,为分子生成、骨架跃迁等任务提供强大工具。主要包括以下几类:
基于语言模型的分子表征
受自然语言处理(NLP)启发,将分子序列(如SMILES、SELFIES)视为特殊化学语言,通过Transformer、BERT等架构处理。例如:
Mol2vec:受Word2vec启发,将分子亚结构视为“单词”,分子视为“句子”,生成密集且信息丰富的向量;
Mol-BERT、MTL-BERT、MolRoPE-BERT:基于BERT框架,通过掩码语言模型任务预训练,捕捉分子的结构和上下文信息,在分子性质预测等任务中表现优异;
MOLFORMER:结合化学SMILES语言和自监督学习,提升分子特征捕捉能力。
这些方法通过令牌化(将原子或亚结构作为令牌)和自监督预训练,学习深层语义关系,为下游任务提供稳健基础,但依赖线性表征,可能无法完全捕捉分子的3D空间和拓扑复杂性。
基于图模型的分子表征
图神经网络将分子表示为图,原子为节点,键为边,能有效捕捉分子的复杂结构细节,适合分子生成和性质预测。例如:
Attentive FP:利用图注意力机制,将节点信息从近邻传播到远邻,捕捉非局部效应;
GROVER:通过自监督学习构建多层次分子图表征,整合消息传递网络和Transformer架构,同时捕捉局部和全局关系;
GraphMVP:在自监督学习中强调2D拓扑和3D几何视图的对齐与一致性,无需显式3D结构信息即可实现稳健的2D分子图编码;
MolCAP:融入化学反应性知识,通过预训练和提示微调,在原子和键层面设计自监督任务,生成高转移性表征。
这些方法能直接建模原子间的结构相互作用,整合几何和层级信息,但存在计算复杂度高、对图构建和消息传递策略敏感等挑战。
基于高维特征的分子表征
整合分子3D结构、结合口袋、细胞表型等高维特征,提升模型表征能力。例如:
UniMol:基于3D分子结构的通用表征学习框架,利用大规模无标记数据预训练,包括约1900万个分子和2.1亿个3D构象;
GeminiMol:引入分子间构象空间相似性概念,通过同一分子编码器生成2048维表征向量,投射到多种分子相似性度量;
PhenoModel:结合化学扰动诱导的细胞形态变化信息,通过对比学习在特征空间中对齐化合物表征与细胞图像,同时捕捉分子构象和潜在活性信息。
这些方法能更好捕捉复杂的空间构象和分子相互作用,提升性质预测和虚拟筛选性能。
多模态分子表征
整合分子图、SMILES序列、指纹、图像等多种模态信息,通过跨模态对比学习或融合框架提升表征的稳健性和可解释性。例如:
FP-GNN:结合分子图信息和分子指纹,通过协同训练进行性质预测;
ImageMol:采用五种预训练策略,将化学知识和结构信息整合到分子图像表征中;
CLAMP、COATI:通过跨模态对比学习,分别对齐化学分子与文本、文本与3D分子表征,生成通用分子嵌入;
VideoMol:利用视觉Transformer从分子视频数据中提取动态和理化信息,实现高精度分子表征。
多模态方法能捕捉分子的结构拓扑、空间构象、序列模式等多方面信息,但有效融合异质数据存在挑战,需复杂的对齐策略减少噪声和冗余。
基于对比学习的分子表征
通过对比正负样本对学习具有判别性的分子嵌入,尤其适用于标记数据有限的场景。例如:
MolCLR:利用GNN对比增强的分子图,生成通用分子表征,在大型化学数据集上预训练,具有良好的可扩展性和迁移性;
iMolCLR:引入加权对比损失解决假阴性问题,在分子和片段层面学习表征;
3DGCL、3D-MOL:专注于3D分子结构,利用SchNet或层级图模型捕捉空间信息,同时保持分子语义一致性。
对比学习能利用大量无标记数据,缓解数据稀缺和类别不平衡问题,但性能高度依赖正负样本对的构建策略,训练稳定性也需关注。
骨架跃迁:从传统到AI驱动的突破
骨架跃迁的定义与重要性
骨架跃迁由Schneider等人于1999年提出,是药物发现和先导化合物优化中的关键策略,旨在发现新的核心结构(骨架),同时保留原分子的生物活性或与靶点的相互作用。其重要性体现在:
(1)改善现有先导化合物的不良性质(如毒性、代谢不稳定性),提升分子活性,优化药代动力学和药效学特征;
(2) 发现结构不同但生物效应相似的新化合物,突破现有专利限制。
骨架跃迁高度依赖有效的分子表征,因为识别保留生物活性的新骨架需要准确捕捉和表示分子的关键特征。
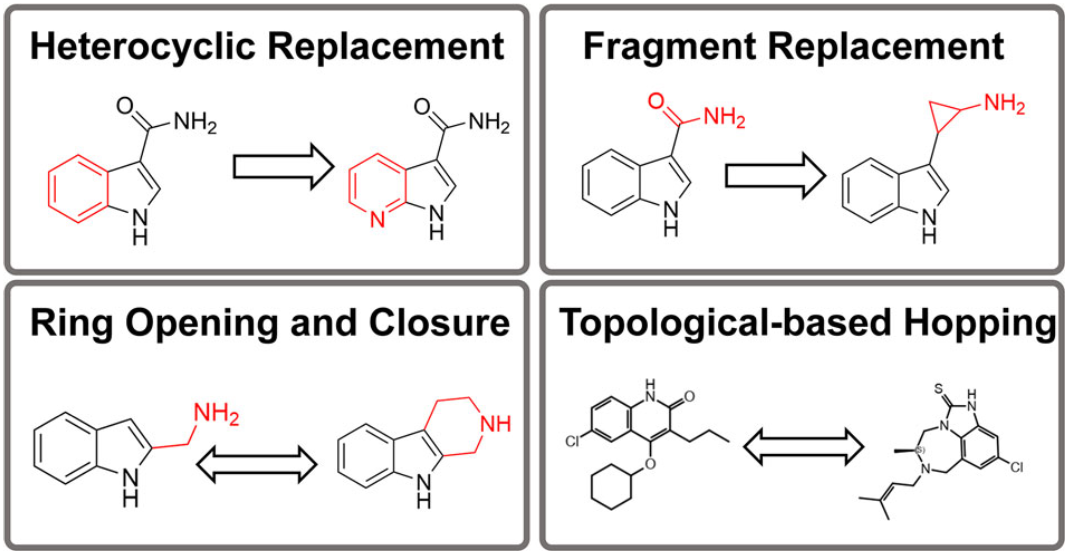
图2 骨架跃迁的典型策略
传统骨架跃迁方法
传统方法主要利用分子指纹和结构相似性搜索,通过替换关键官能团(如保持氢键模式、疏水相互作用、静电力等关键相互作用),同时引入新的分子片段结构,识别具有相似性质但核心结构不同的化合物。例如:
pharmacophore modeling:表示生物活性关键分子特征的空间排列,如NScaffold利用拓扑药效团图(PhGs)编码药效团特征;
形状相似性方法:如ROCS通过形状叠加检测具有相似结合性质的分子体积,SHAFTS结合形状叠加评分和药效团特征匹配,提升骨架发现效率;
指纹和相似性搜索:如ErG将分子图抽象为具有药效团类型节点的简化图,WHALES整合几何、原子距离和分子性质信息,提供全面的3D表征。
但传统方法依赖预定义规则、固定特征或专家知识,探索化学空间的能力有限。
现代AI驱动的骨架跃迁方法
基于深度学习的现代方法通过数据驱动的灵活探索,极大扩展了骨架跃迁的潜力,尤其生成式模型能设计全新骨架。主要包括:
基于图和VAE的生成模型
通过解耦分子组件,实现精确的骨架修饰。例如:
GraphGMVAE:采用高斯混合变分自编码器,将骨架和侧链编码为独立分布,在JAK1抑制剂上验证,生成新骨架的成功率达97.9%,部分合成化合物具有强生物活性(IC50为5.0nM);
ScaffoldGVAE:结合多视图图神经网络,捕捉骨架层面和分子动力学特征,在多种激酶靶点上成功率接近100%,成功生成高活性的新型LRRK2抑制剂。
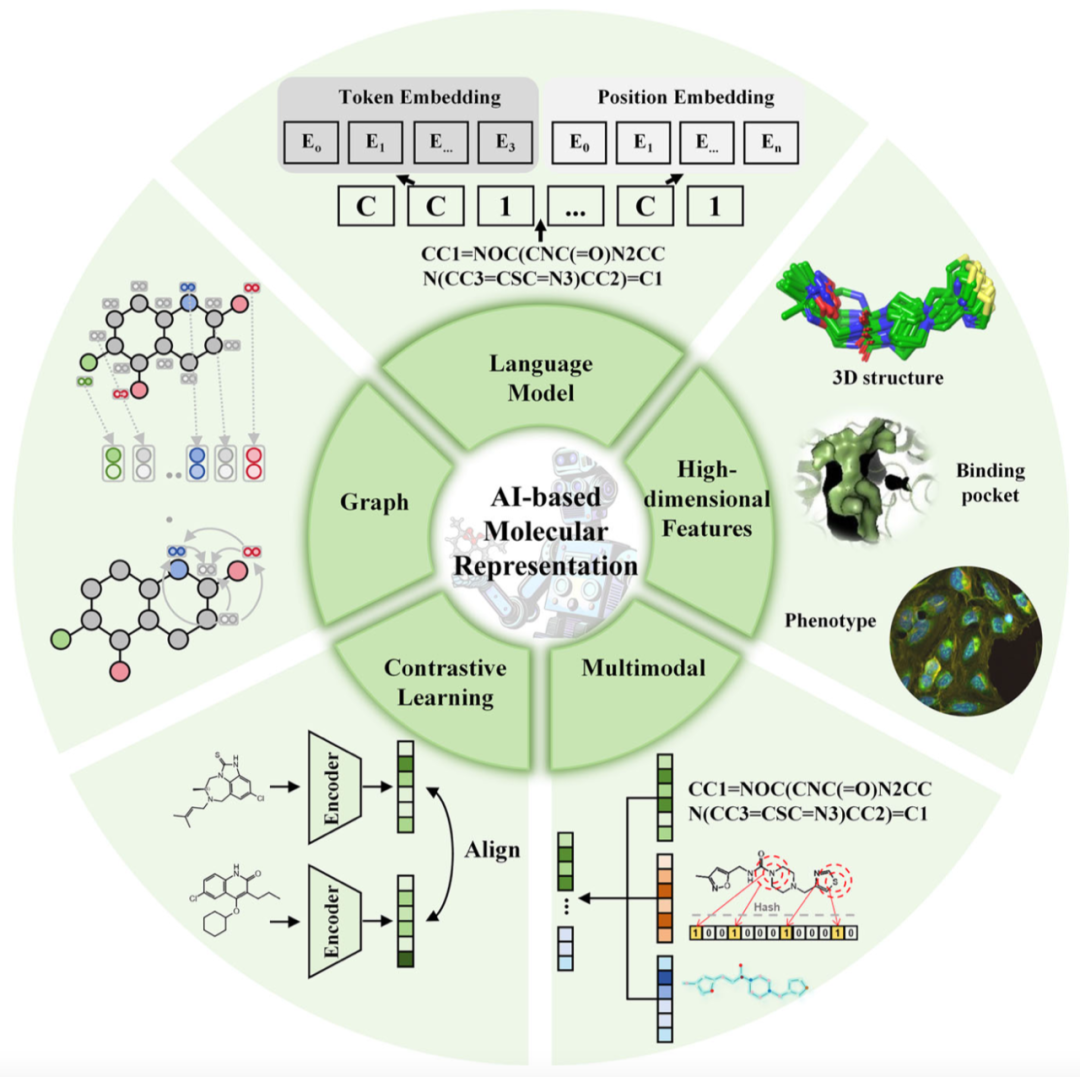
图3 五种主要的基于人工智能的分子表征模型示意图
多模态和药效团引导方法
整合多种分子数据确保生物活性。例如:
DeepHop:结合3D分子结构和蛋白质序列嵌入,利用Transformer架构优化骨架生成,在57,000多个骨架跃迁对上训练,成功率达65.2%;
PGMG:将药效团特征编码为完整图,处理药效团与分子的多对多关系,生成具有增强生物活性和类药性的新型EGFR抑制剂。
基于扩散模型的方法
提供精确的空间控制,提升骨架多样性和结合亲和力。例如:
DiffHopp:采用E(3)等变图扩散模型,结合几何向量感知器(GVP)编码器和3D分子图扩散,在PDBBind数据集上的QED、SA、Vina评分等指标表现优异;
DiffSBDD:整合蛋白质口袋信息,实现上下文感知的骨架跃迁,优化结合亲和力等分子性质,在Binding MOAD数据集上的对接分数和合成可及性优于基线方法;
TurboHopp:引入E(3)等变一致性模型和强化学习,将骨架生成速度提升30倍,同时改善分子的连接性、结合亲和力和可合成性。
其他创新方法
REINVENT 4:整合强化学习、迁移学习和课程学习,实现多样化的骨架修饰,生成创新连接子和新核心骨架;
Ouroboros:在预训练分子编码器的 latent空间中采用定向化学进化策略,通过迭代“突变”和选择过程,引导分子表征从一个骨架区域过渡到另一个,实现可控的骨架跃迁。
挑战与展望
尽管分子表征和骨架跃迁方法取得显著进展,仍面临诸多挑战:
过度强调基准性能
多数方法主要在标准基准数据集上评估性能,陷入“排行榜竞赛”,而忽视实际科学挑战。部分研究表明,通过有效结合分子图信息与分子指纹特征,合理选择模型架构和参数调优,传统表征方法也能达到高性能。未来需结合AI和传统方法,开发通用、自适应的AI架构,实现对不同基准场景的自调整。
依赖数据质量和数量
AI方法严重依赖训练数据的质量和数量,数据集不足、实验数据的批次效应、标签偏差等会影响模型性能。高质量标记数据获取昂贵,大型实验数据集多为商业机构专有,限制模型的普适性。解决策略包括:
(1) 联邦学习:多机构协同训练,不直接共享敏感数据,通过聚合模型更新保护隐私并利用多样化数据集;
(2) 联邦学习与知识蒸馏结合:如DeepSeek等大语言模型的做法,将复杂的本地模型(教师)知识转移到紧凑的全局模型(学生),提升模型稳健性。
化学空间探索的局限
传统方法受预定义规则限制,现代AI方法也可能因训练数据集分布导致生成的骨架收敛于特定化学型,降低多样性。潜在解决方案是采用基于注意力的多模态融合网络,自适应学习2D、3D和深度学习表征之间的关系,通过专用融合层整合多模态特征,提升化学空间探索效率和骨架跃迁成功率。
合成可及性和类药性
AI生成模型虽能创建新骨架,但确保其合成可行性和类药性仍具挑战,许多生成分子可能难以合成或药代动力学/药效学特征不佳,需额外筛选。解决方案包括在生成模型的奖励函数中整合合成可及性评分、功能反应模板或逆合成预测算法,优先生成新颖且可合成的分子。
多模态表征整合的挑战
多模态分子表征模型虽增多,但有效整合2D、3D和深度学习表征到药物发现流程中仍存困难,且模型的可解释性差,难以理解特定分子特征如何影响预测活性,阻碍分子的合理优化。
结语
分子表征方法的快速演进,尤其是AI驱动的创新,为药物发现中的骨架跃迁提供了强大工具,从传统的规则驱动方法迈向数据驱动的灵活探索,极大扩展了化学空间的探索范围。尽管面临数据、化学空间探索、合成可及性等挑战,但通过跨学科合作和技术创新,这些方法有望加速新药研发,为疾病治疗带来新的突破。未来,随着模型稳健性、可解释性和实用性的提升,AI在药物发现中的作用将更加不可或缺。
参考资料
Wang, S., Zhang, R., Li, X. et al. Recent advances in molecular representation methods and their applications in scaffold hopping. npj Drug Discov. 2, 14 (2025).
https://doi.org/10.1038/s44386-025-00017-2
--------- End ---------
内容中包含的图片若涉及版权问题,请及时与我们联系删除
