
Published on July 14, 2025 5:32 PM GMT
TL;DR: The Gram matrix of the normalized activations – viewed as an image – is surprisingly useful for revealing the structure of activations in protein language models. A number of examples are presented.
Purpose: Trial balloon for a longer post on a much larger project.
I'm an independent AI-for-bio researcher based in Boston. If you’re interested in this research and in the area, reach out – I’d love to grab coffee.
For the past several weeks I have been focused on creating a DNA language model that is more suited to mechanistic interpretability analysis than current transformer/SSM-based models. That project has seen some success, and a report on it is forthcoming.
This post is not about that project, but a tool I’ve been using to analyze the model. I want to understand what this community thinks about the technique and the results it produces before I perform more analyses with it.
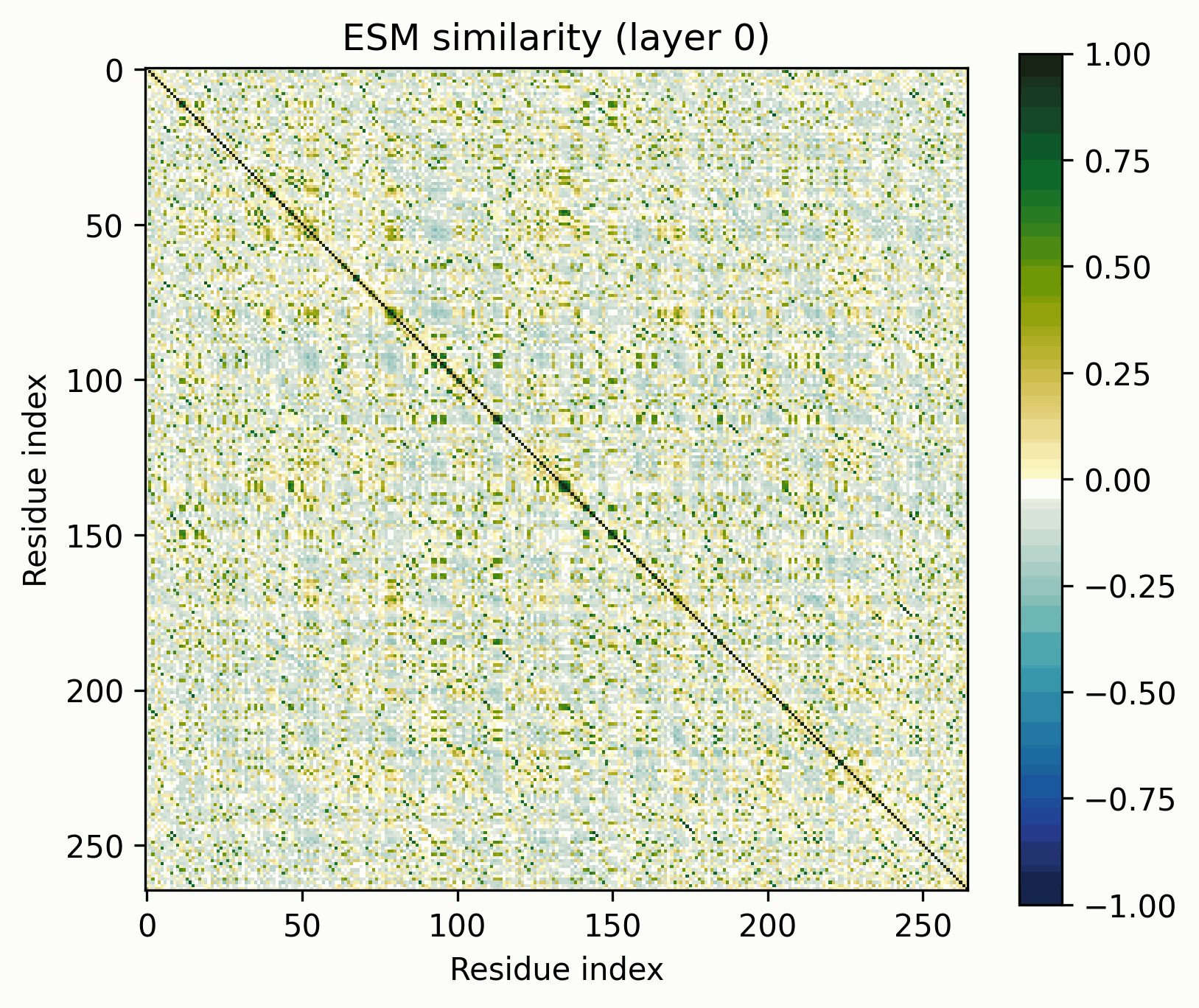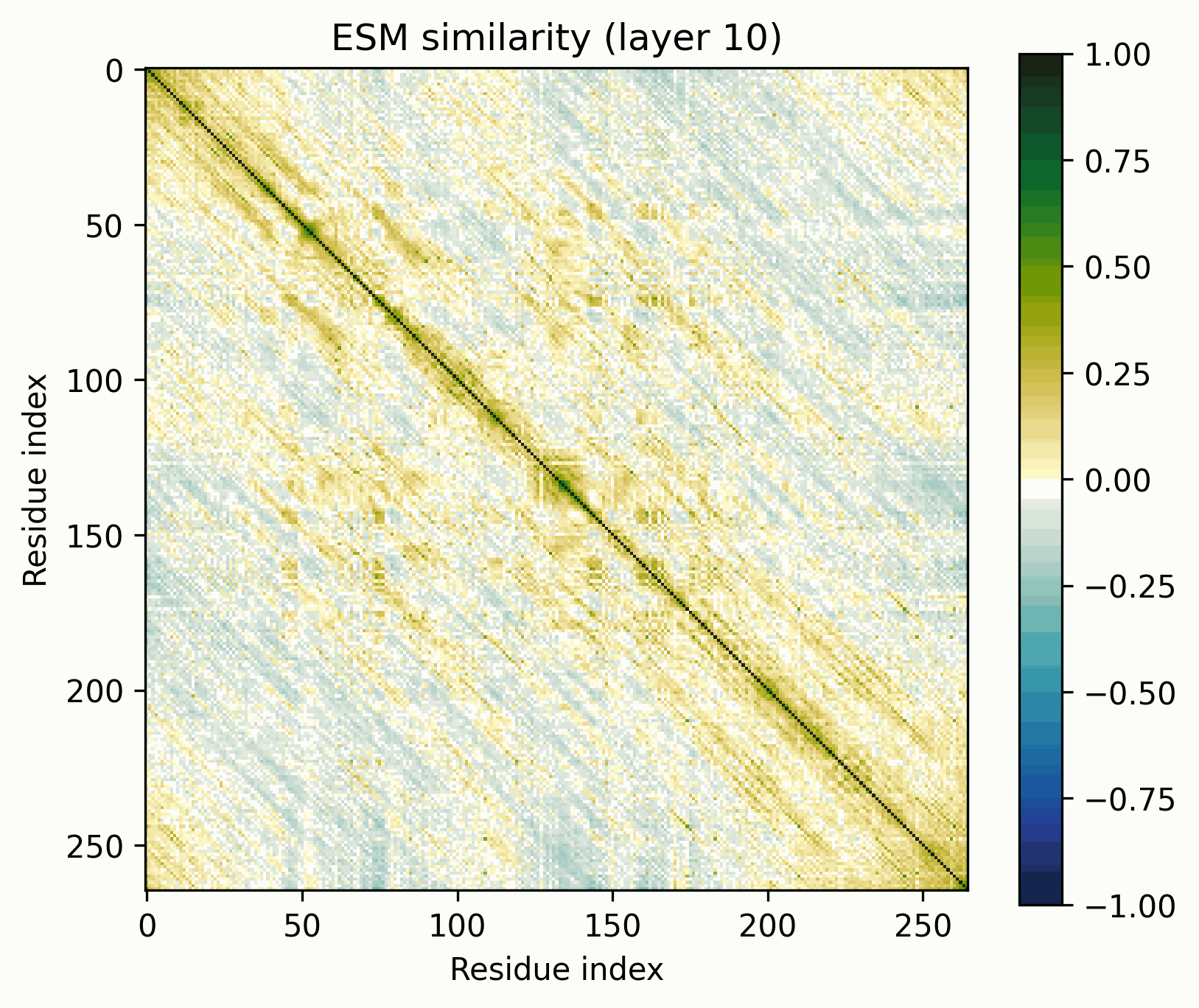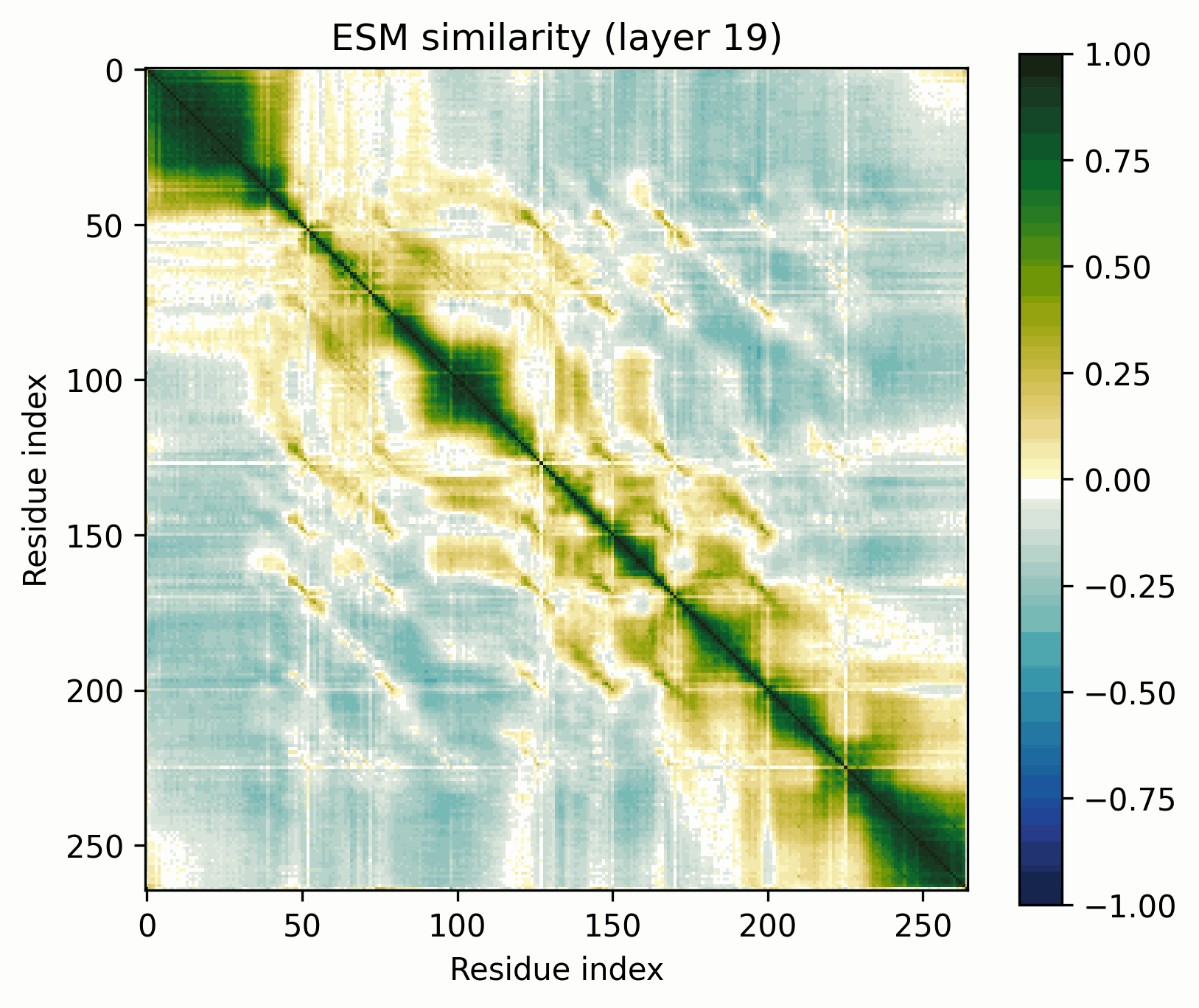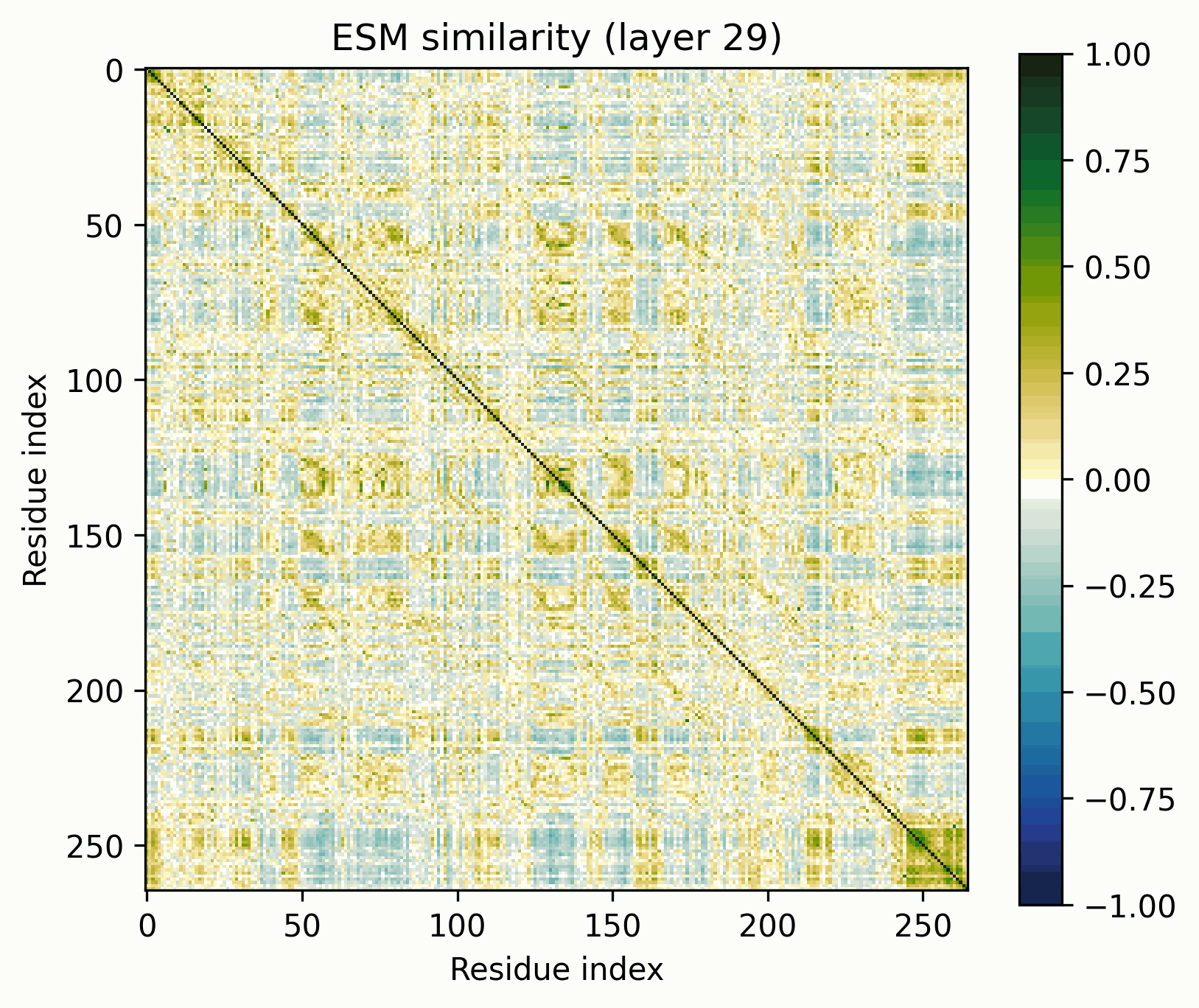
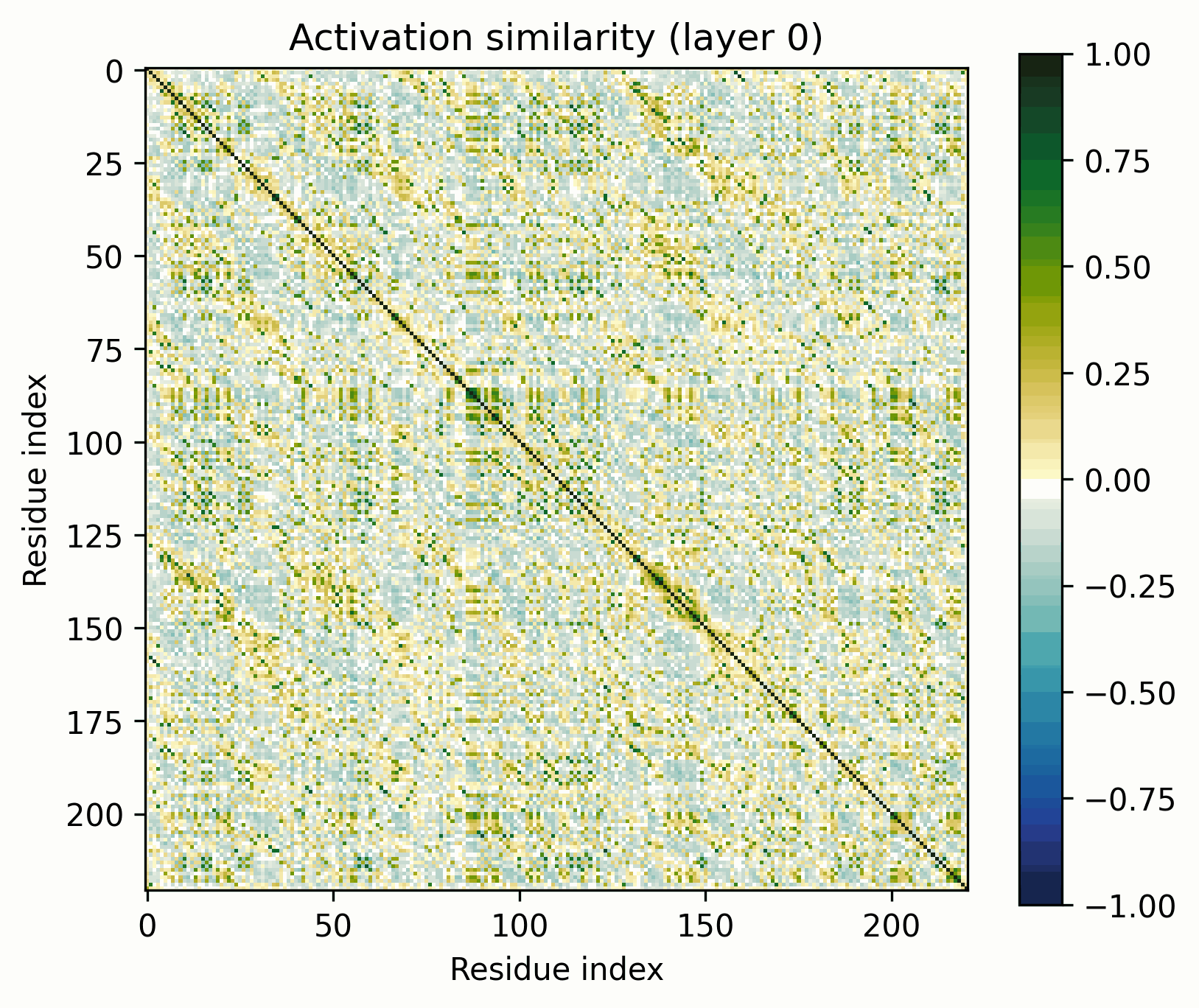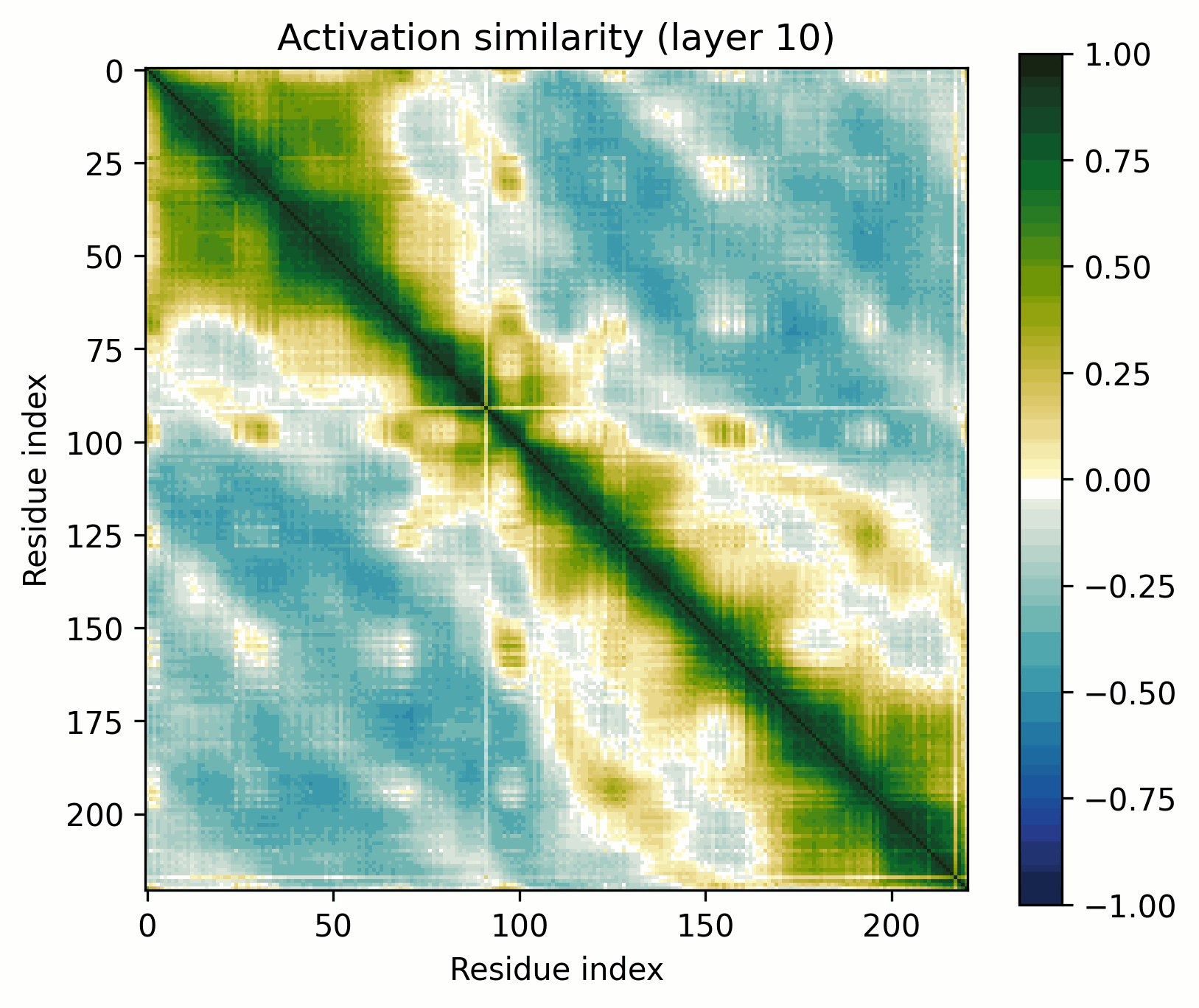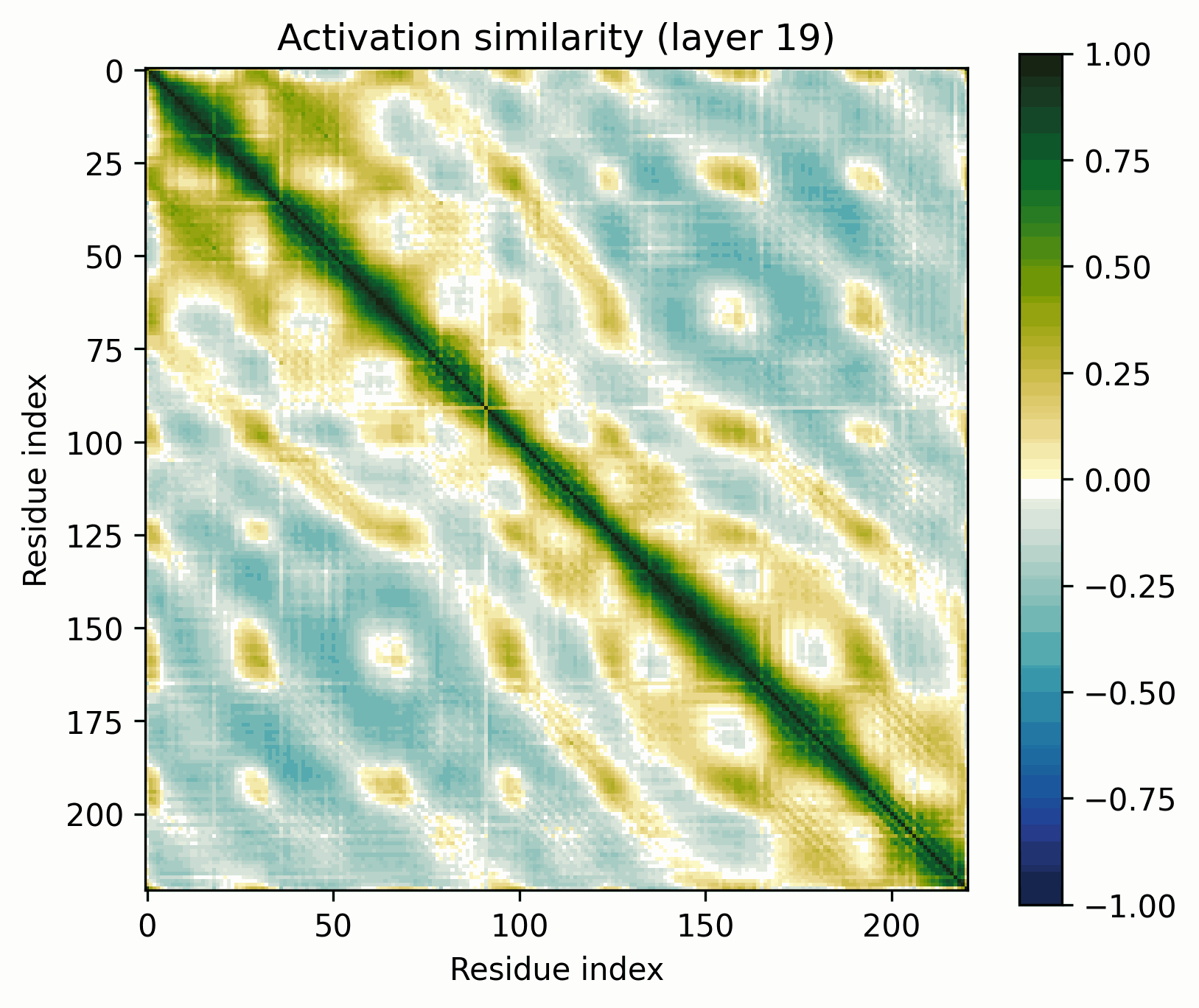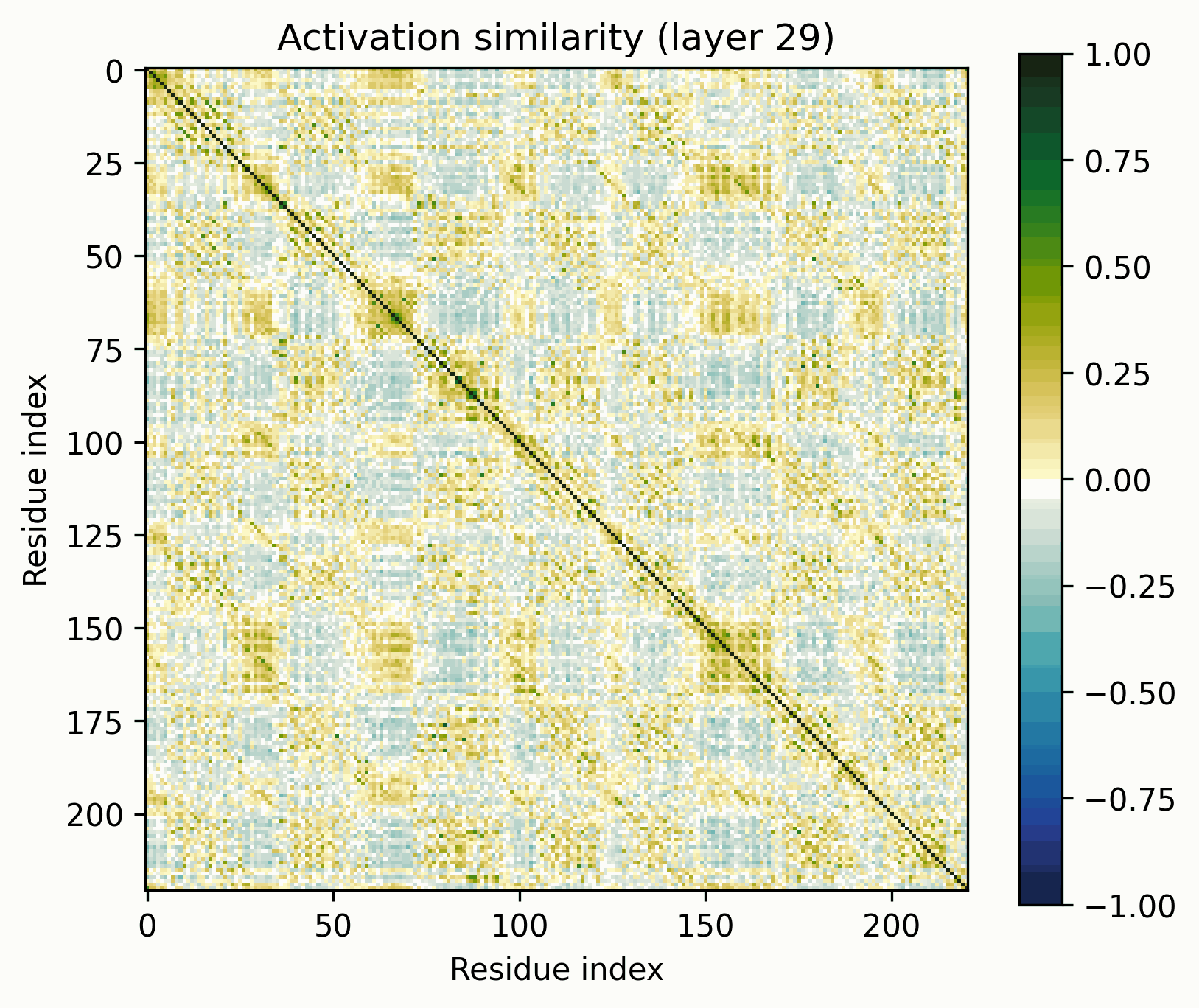
The tool is simple: take a set of activations across a sequence, mean-center them, normalize to unit magnitude, and visualize their pairwise similarities (i.e., the Gram matrix). The resulting images are visually striking and surprisingly useful for revealing the structure of the activations.
I won’t be analyzing my DNA language model in this post, but rather the ESM-C 300M protein model from EvolutionaryScale. I chose ESM-C to analyze because it is an openly available transformer-based model from a frontier AI-for-bio lab with extensive tooling.
These models differ from my model in training objective (MLM vs autoregressive), domain (proteins vs DNA), and architecture. However, the takeaway is the same: biological language models contain unusual features that SAEs might not reliably or completely capture.
Why the Gram matrix? Why biological LLMs?
I’ve only found a few examples of this method being used in the past: here, and here for positional embeddings. I don’t believe it has a name: following the “logit lens,” I propose that it be called the Gram lens.
A biological sequence can be segmented in a way that language cannot: into operons, ORFs, folds, beta strands, alpha helices, etc. There ought to be swaths of off-diagonal similarity if the network has learned these concepts, wherever two parts of the sequence have the same quality. This is largely why we’d expect the Gram lens to be more helpful with biology than with language.
I have a lot more to say (aimed at an audience of biologists) about why the current paradigm of mechanistic interpretability is likely to be important and useful for biology, whatever its utility for alignment. That's coming with the next post. Briefly: if you’re in alignment, why should you care?
- Biological sequences provide a natural testbed for interpretability techniques meant for language models. They share the same string-of-tokens format as natural language, but have a completely different underlying structure.Interpretability techniques developed for language models will be optimized until their failures are edge cases. Biology is sufficiently distinct from language that it might surface these edge cases.Insofar as biological capabilities in themselves constitute a meaningful risk from AI, then interpretability may be a useful tool for detection and mitigation.Linear probes on model latents are already a major tool of the trade in the protein engineering industry, and they have been for years. If your method provides a gain over linear probes in terms of interpretability or (especially) generalization in practice, there is money and glory available in wet-lab-validated protein engineering competitions. Sparse linear probes on SAE latents are themselves promising.
Note on Interpretation
Before we get started, let’s clarify what these images do and do not represent. They show the similarity of the normalized activations between every pair of amino acids. To the extent that the directions of the activations determine their meaning, this geometric similarity mirrors their semantic similarity. While they are visually similar to contact maps and categorical Jacobians, they do not necessarily represent the same information.
For example, we might expect two alpha helices on opposite ends of a protein to show similarity, even if they are neither in contact nor strongly coevolve. This would simply be because they share the quality of being alpha helices. We could even see this similarity between entirely separate proteins, each with alpha helices, if we were to compare cross-protein activation similarities.
However, protein language models must grasp coevolutionary relationships/close contacts to make accurate predictions. Because attention heads represent similarities within subspaces of the residual, these images may well occasionally resemble contact maps.
What will these images look like?
A note on wording: I have seen the term “features” being used to refer to at least three different things, which has frustrated me to no end. For the purposes of this post, I’m going to (try to) stick to this terminology[1]:
- A primitive is a class of structure into which the activations can be decomposed. For example, “directions considered independently across positions” or “a sinusoid across positions” might be primitives.A feature is an instance of a primitive.A concept is a singular unit of meaning. This is inherently subjective. If we determine that a feature corresponds to a concept, we have found a monosemantic feature.
Let’s look at some potential primitives to get a sense of what the images we’ll see might represent. Consider a linear feature with a binary on/off activation. Research with sparse autoencoders often treats these features as "natural" in some sense.
If a feature like this is activated at multiple positions in the sequence, we will see a grid.
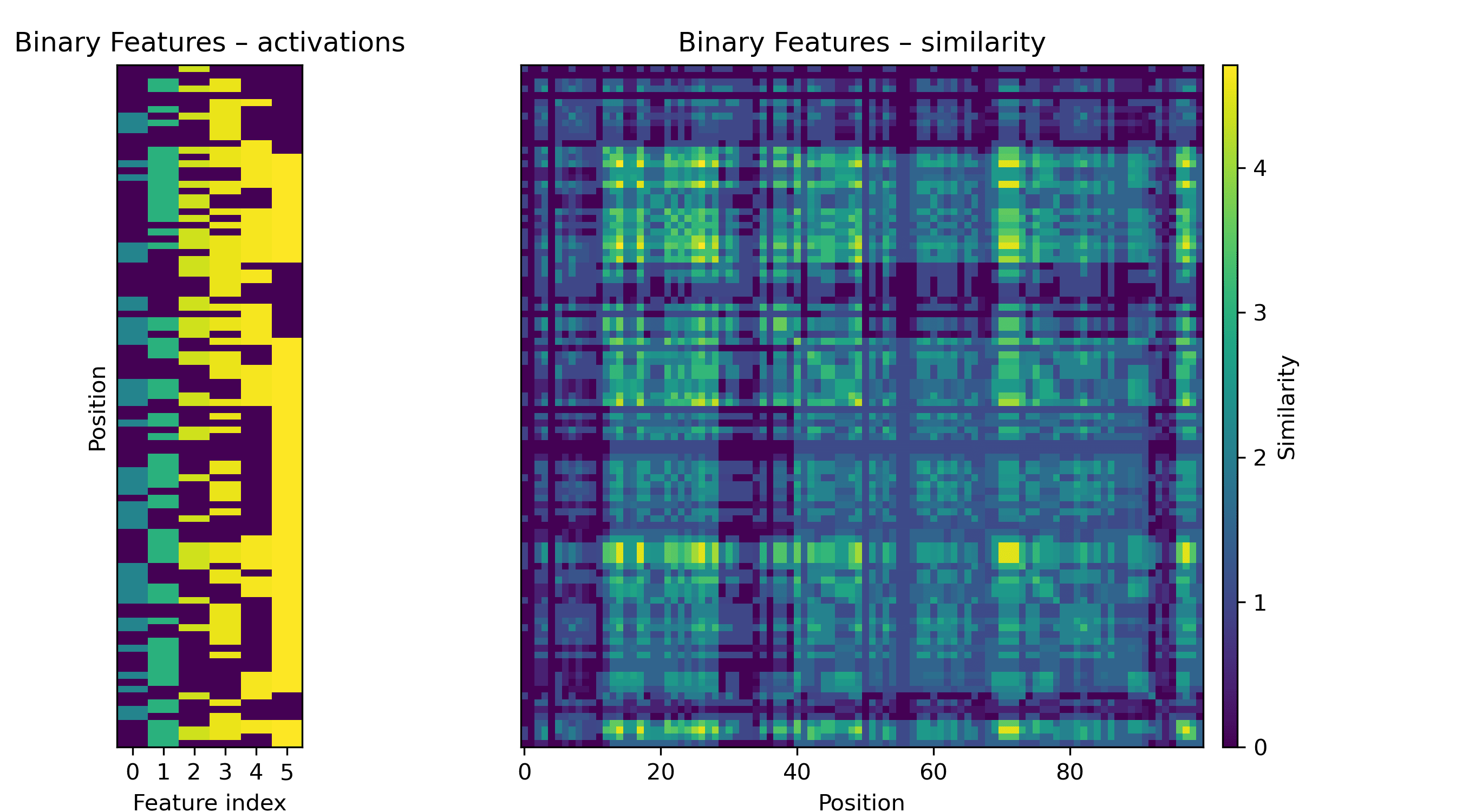
If the activation is a sinusoid, we will see a fuzzy checkerboard.
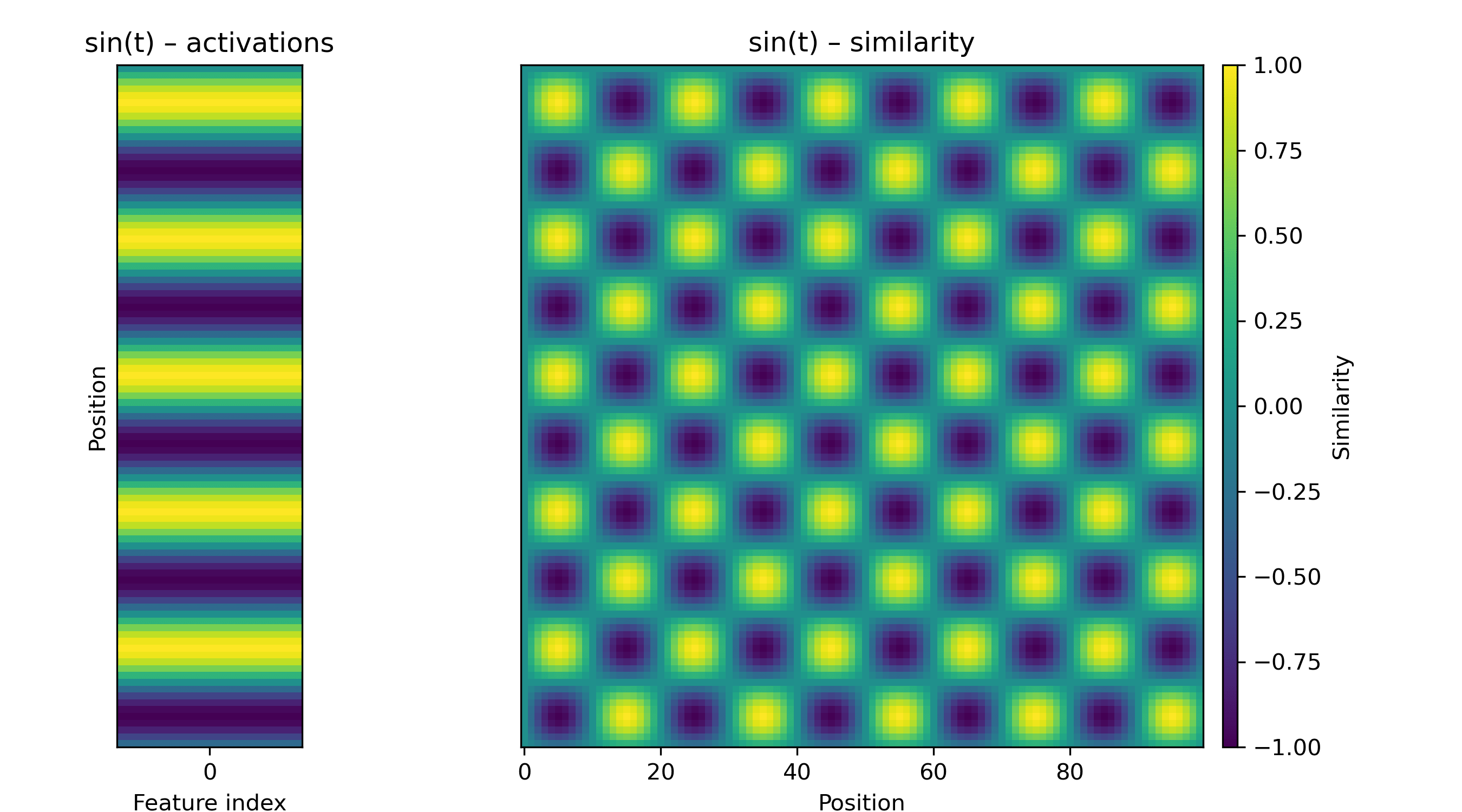
Another pattern we might encounter is a repeating diagonal, which corresponds to a set of periodically active features. We can make a blurry one with a paired-sine-and-cosine feature, i.e. a rotation in a 2D subspace:
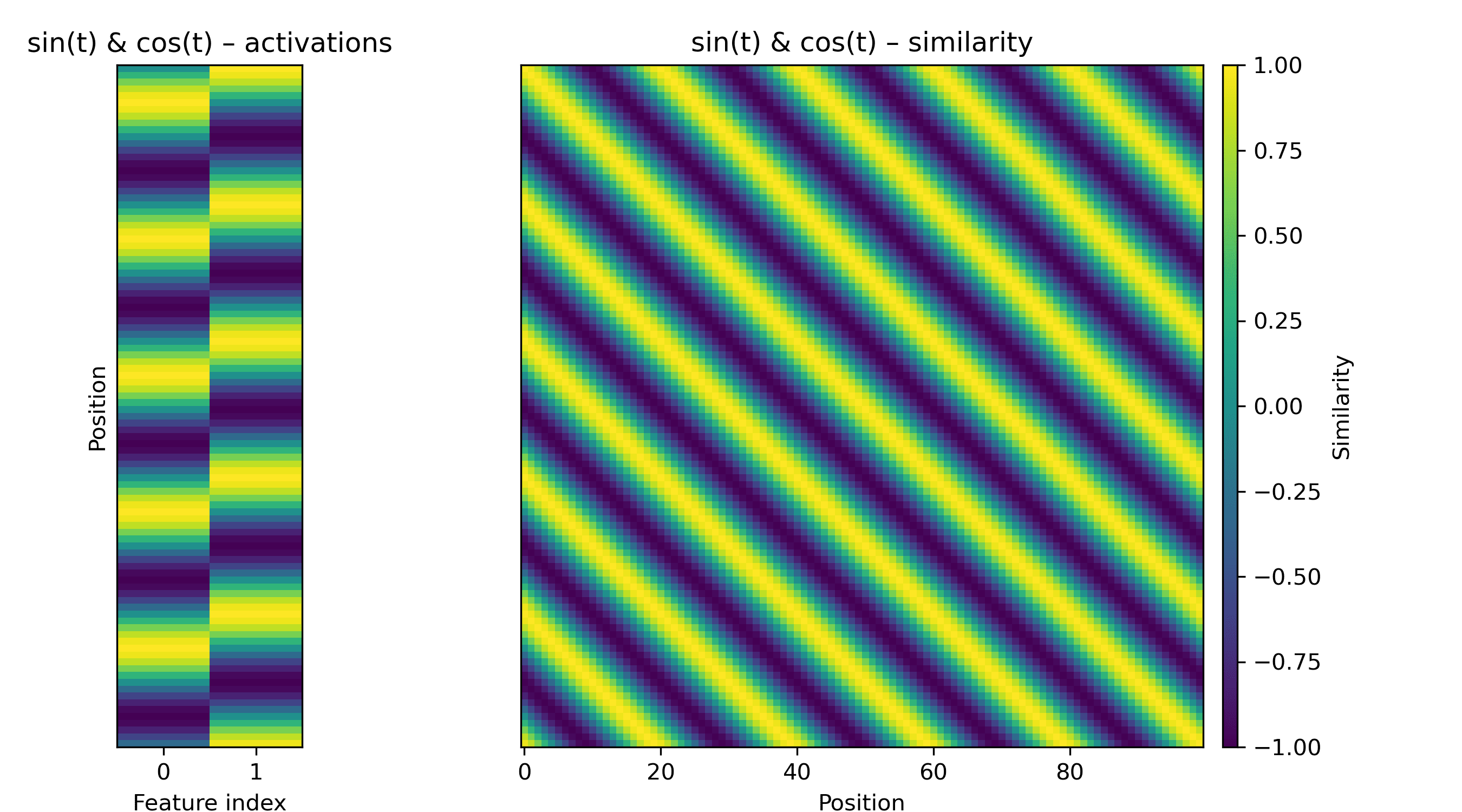
We might also see sharper diagonals. These are somewhat space-intensive: for a repeat every, say, 7 amino acids, we need seven orthogonal directions if the off-diagonals are zero.
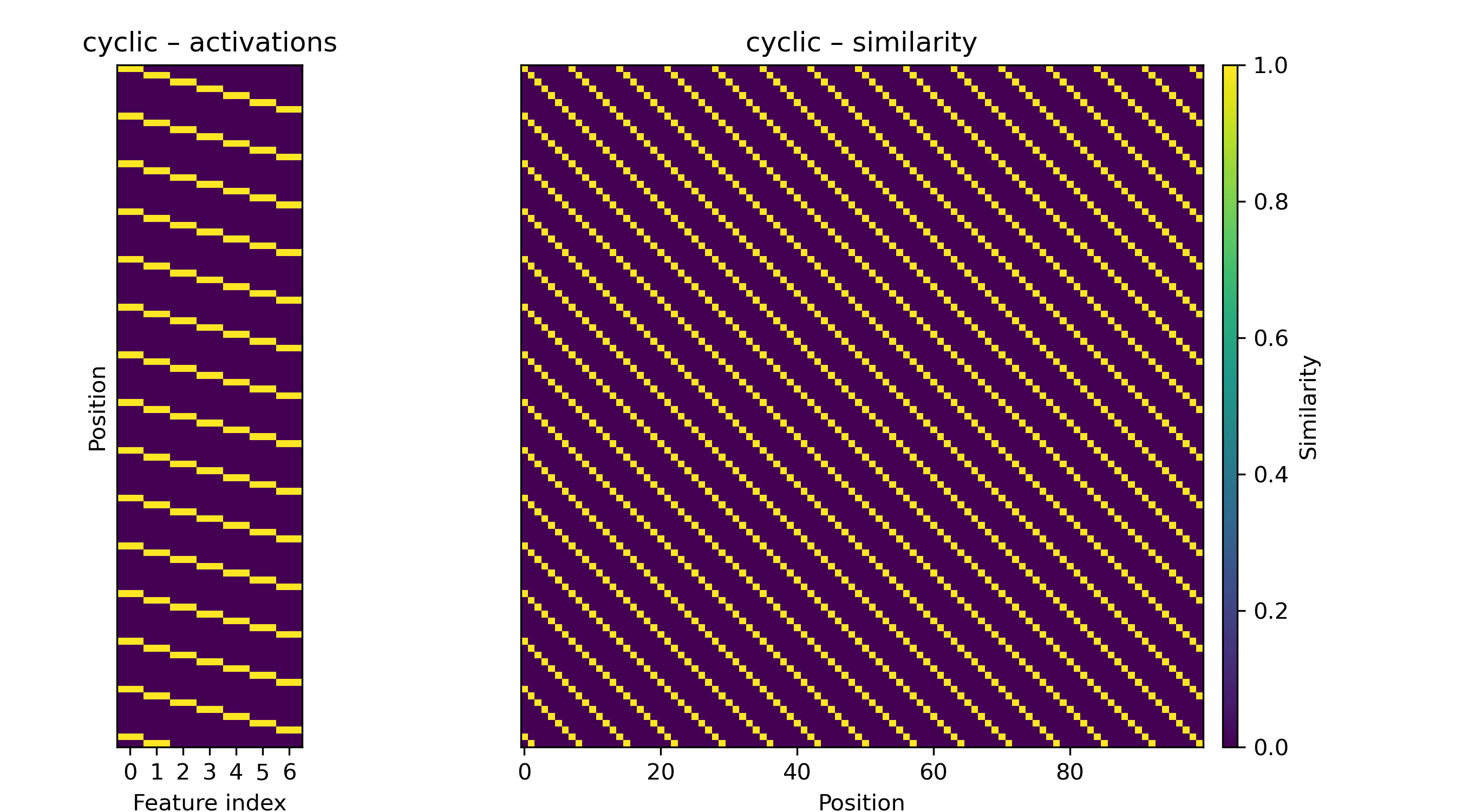
A non-repeating diagonal, be it the identity matrix or a matrix with multiple diagonals, is much more space-intensive. Such a matrix is full-rank, and low-rank approximations are difficult to construct without a nonlinearity on the similarities.
We can anticipate more exotic primitives, too. The attention heads of protein language models are known to approximate whether two positions contact each other. This might require something like a low-rank approximation of the distance matrix in the residual stream.
It’s important to note that any of these “features” might reflect the structure of the input rather than the workings of the model per se. Consider a toy model that assigns each 6-amino-acid segment to a random, high-dimensional unit-norm vector. This model has not learned anything meaningful.
On an ordinary protein, the activations of this model would produce a close approximation to the identity when viewed via the Gram lens: the random vectors are all nearly orthogonal, and 6-mers are unlikely to occur twice in an arbitrary protein. However, a protein composed of two repeated identical domains would have an additional offset diagonal. This is an interpretable and faithful representation of the underlying protein, but has little to do with what the model has “learned.”
Some limitations of the method: there are many circumstances where a feature’s presence will not be made clearer by the Gram lens, notably features that appear at every position in the protein or have very small maximum magnitudes. It's also not possible to faithfully infer the underlying features that produce a map, though it usually provides strong hints.
Examples
To keep these findings easy to replicate, I’m going to be using the return_hidden_states functionality that EvolutionaryScale provides for ESM-C. This returns the residual stream after every transformer+feedforward block. The activations after "layer n" mean after the nth block in this case.
Alpha Helix
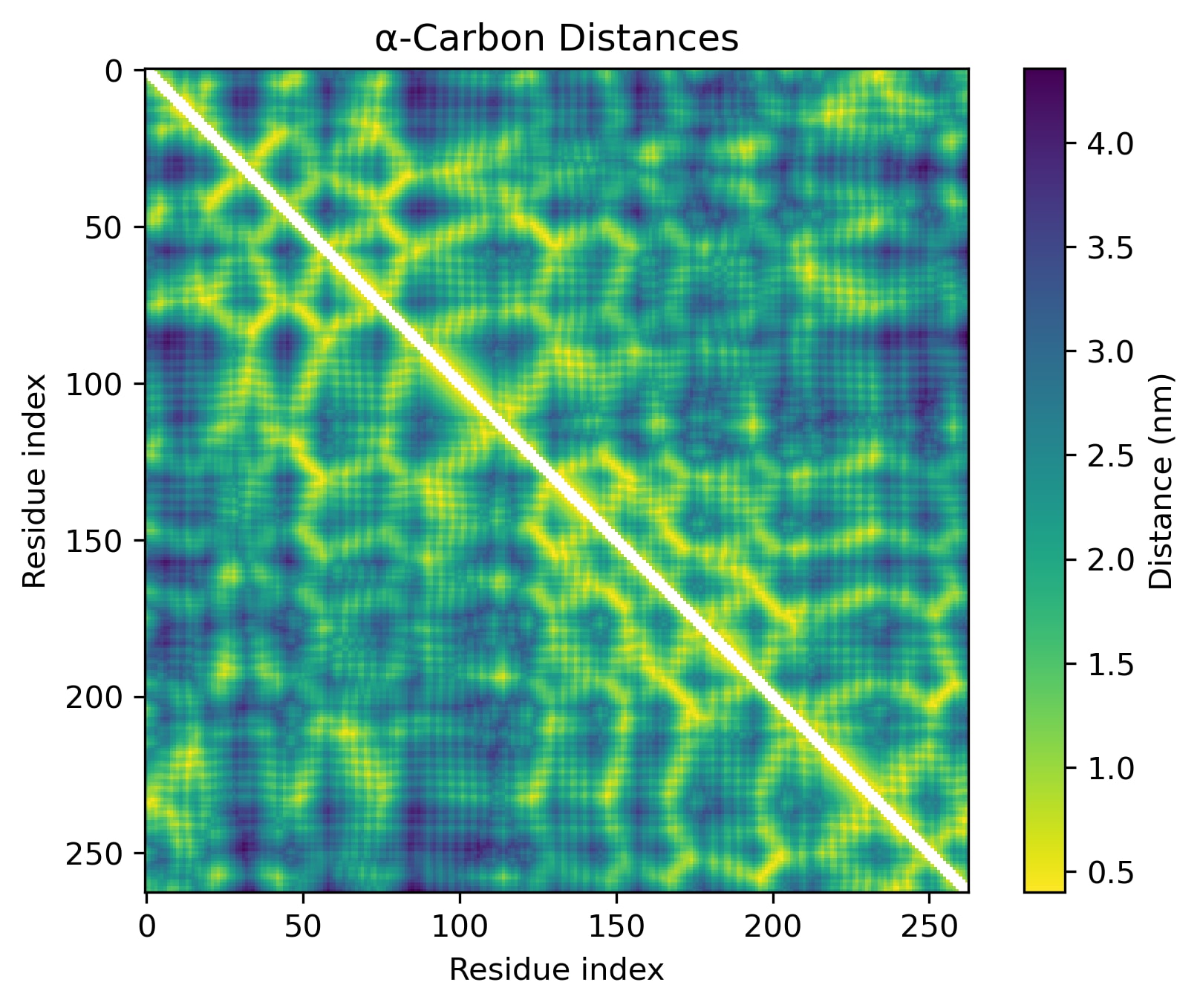
Let’s look at some proteins to see what they’re like. Protein Q8VD04, which I got from InterProt, is a good place to start – it’s mostly just some long alpha helices, so we might get[2] a clean view of what an alpha helix looks like in isolation.
Here's that last segment, zoomed in:
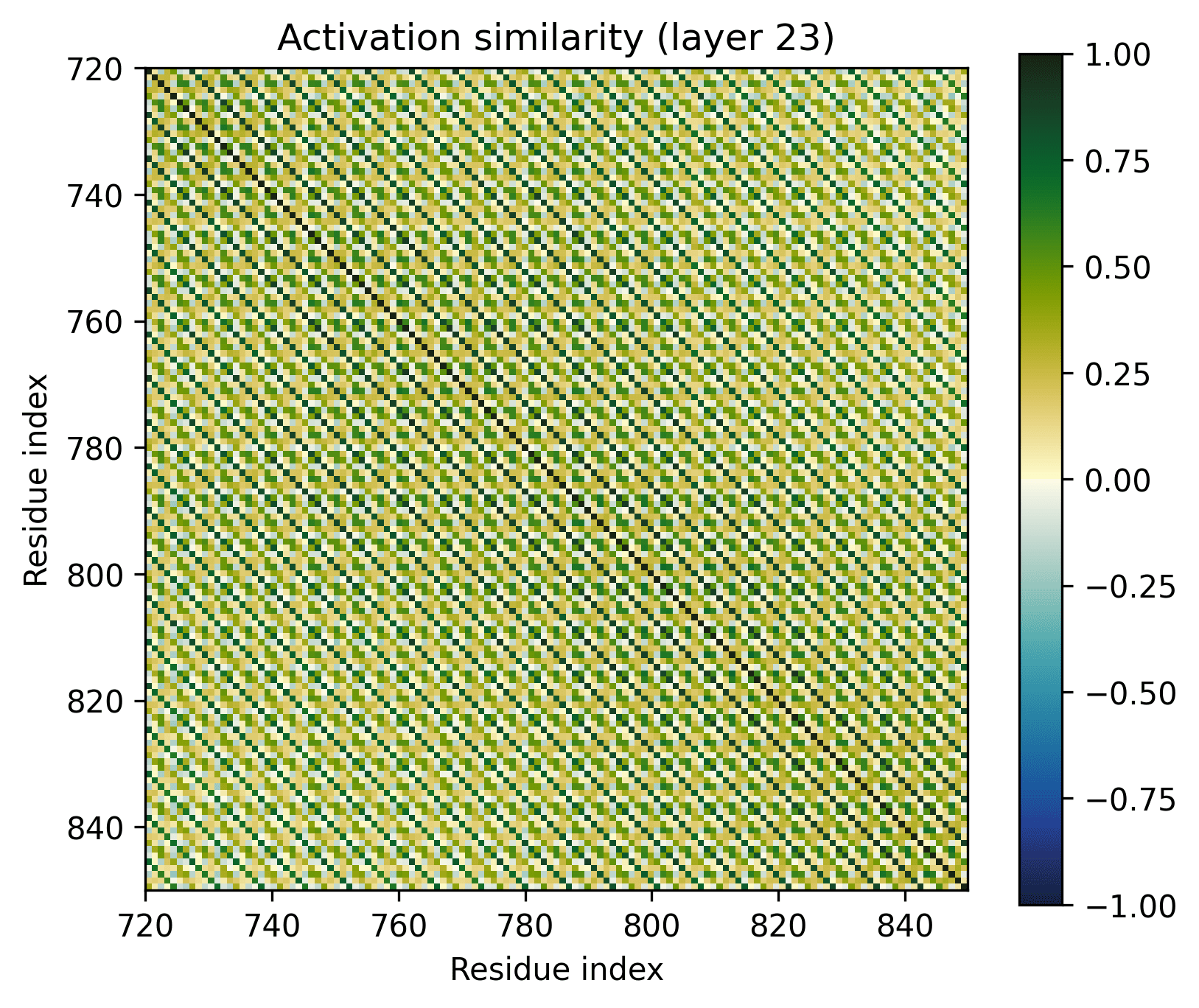
Every seventh amino acid clearly has a distinct representation, judging by the diagonal pattern. However, these representations are obviously not at all orthogonal. An alpha helix appears to be defined by this pattern per se, rather than by a linear feature.
What would these features look like through a SAE? InterProt (which, it should be noted, analyzed a different model: ESM-2-650M) found some relevant features.
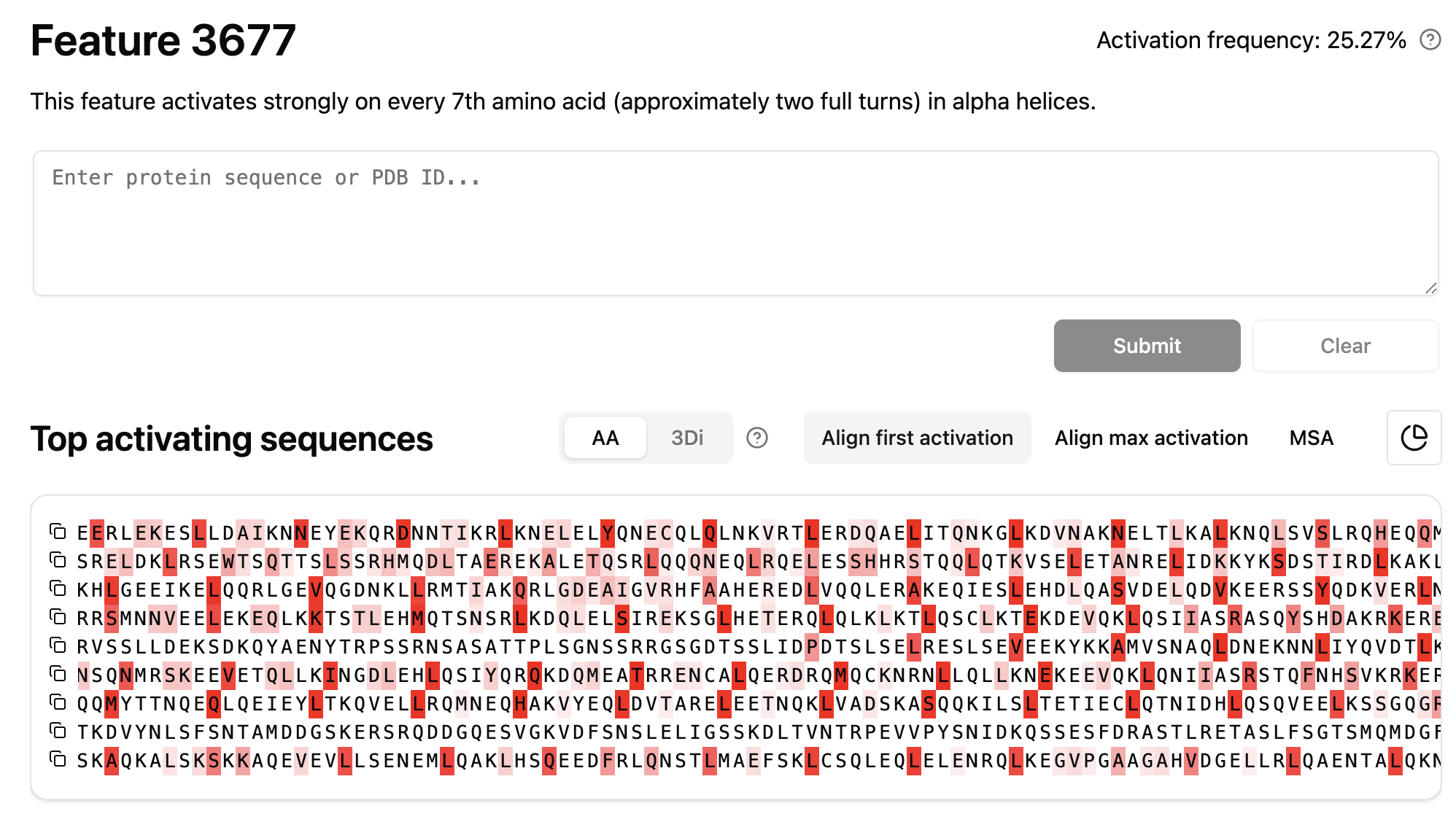
The correspondence to the Gram lens is obvious. However, to identify the complete pattern with SAE features alone, you would have to find every relevant feature. The Gram lens reveals the overall pattern, but cannot reliably show how the pattern was originally represented. The SAE features are likely to be helpful to identify the underlying structure, so the methods are complementary.
Within a long helix, and from one end of the protein to another, the similarity slowly diminishes. A major outstanding question is why?
One possibility is that activations can be decomposed into the sum of a pure alpha-helix-representing component and a positional component. A SAE could plausibly separate these. Another possibility is that the subspace containing the alpha helix representations slowly rotates across the sequence. If the latter is true, and the effect is common, this might substantially hinder the interpretability of bio model SAEs.
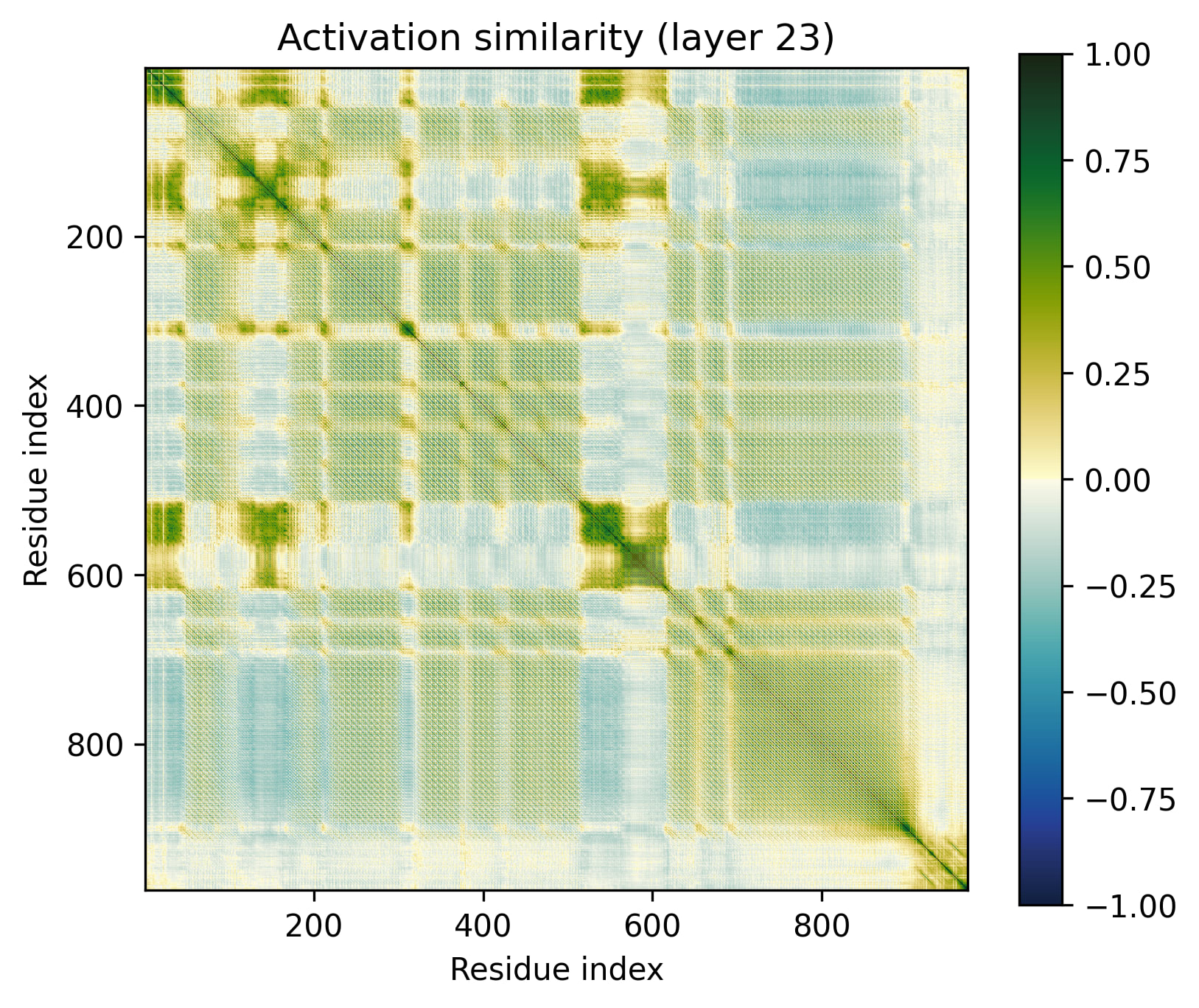
Alpha Barrel
We can up the complexity a bit by going from alpha helices to a transmembrane alpha-helical barrel. Bacteriorhodopsin is a good example (Uniprot: P02945, PDB: 1AP9).
Layer 24 seems interesting, let's take a closer look.
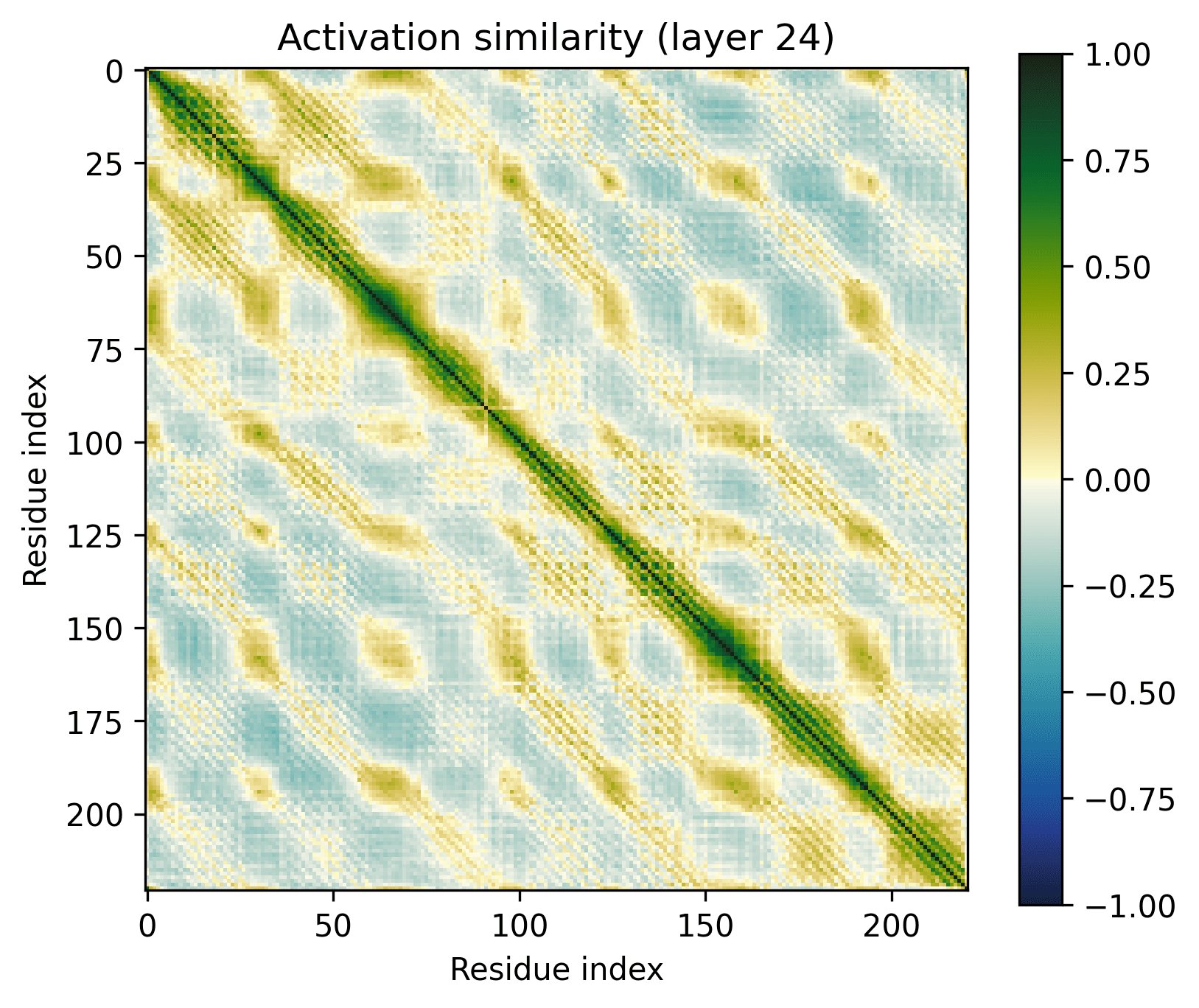
Observations:
- There's a clear division between the helices and the connecting loops.The overarching pattern is grid-like: roughly what we’d expect from a single sine-like feature. Alternatively, they could be represented independently, with the oscillation reflecting the repetitive organization of the protein.The helices have a similar pattern to the long alpha helices overlaid on them: diagonal stripes and a grid[3].Overall similarity decays somewhat with distance, though there is often a greater degree of similarity between every second helix/loop. These share an orientation (direction for helices, side of the protein for loops).The near-diagonal stripe is quite substantially darker than the rest of the image.
Note the patterns in the early layers, starting with layer 1. As previously mentioned, the 1D features that SAEs consider form grids on the off-diagonal. Diagonals are not compatible with features that SAEs could independently resolve.
Diagonals are everywhere in these layers:
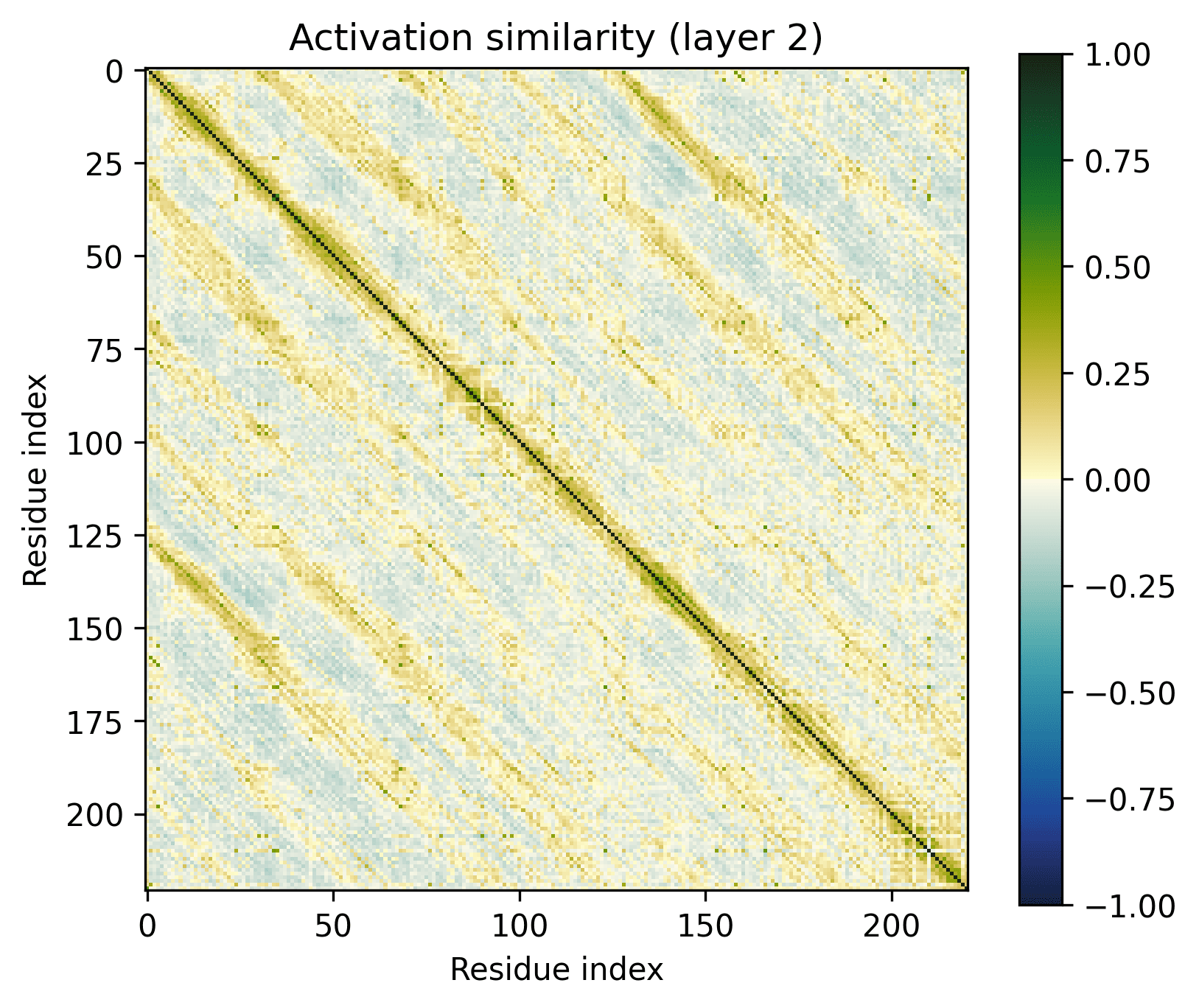
Layers dominated by this pattern of activation are ubiquitous in the early layers of the biological sequence models I've investigated, across modalities and architectures. I'm not entirely sure what kind of primitive would produce this pattern of activation (sparsely activated sinusoidal features?), but I don't see how it could be SAE-compatible.
MFS Transporter
The ubiquitous Major Facilitator Superfamily of transporters are composed of two repeating domains, themselves composed of three alpha helices.
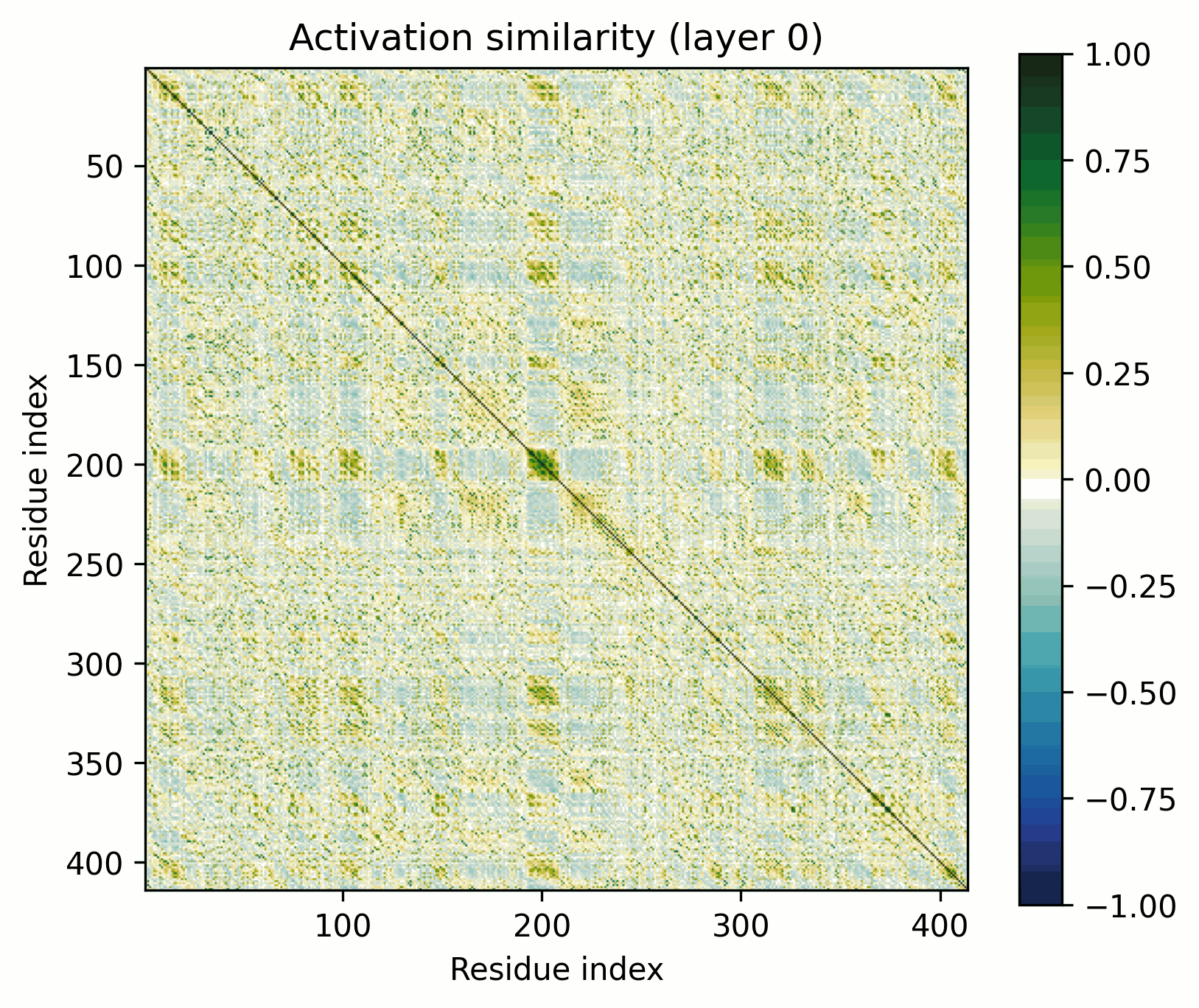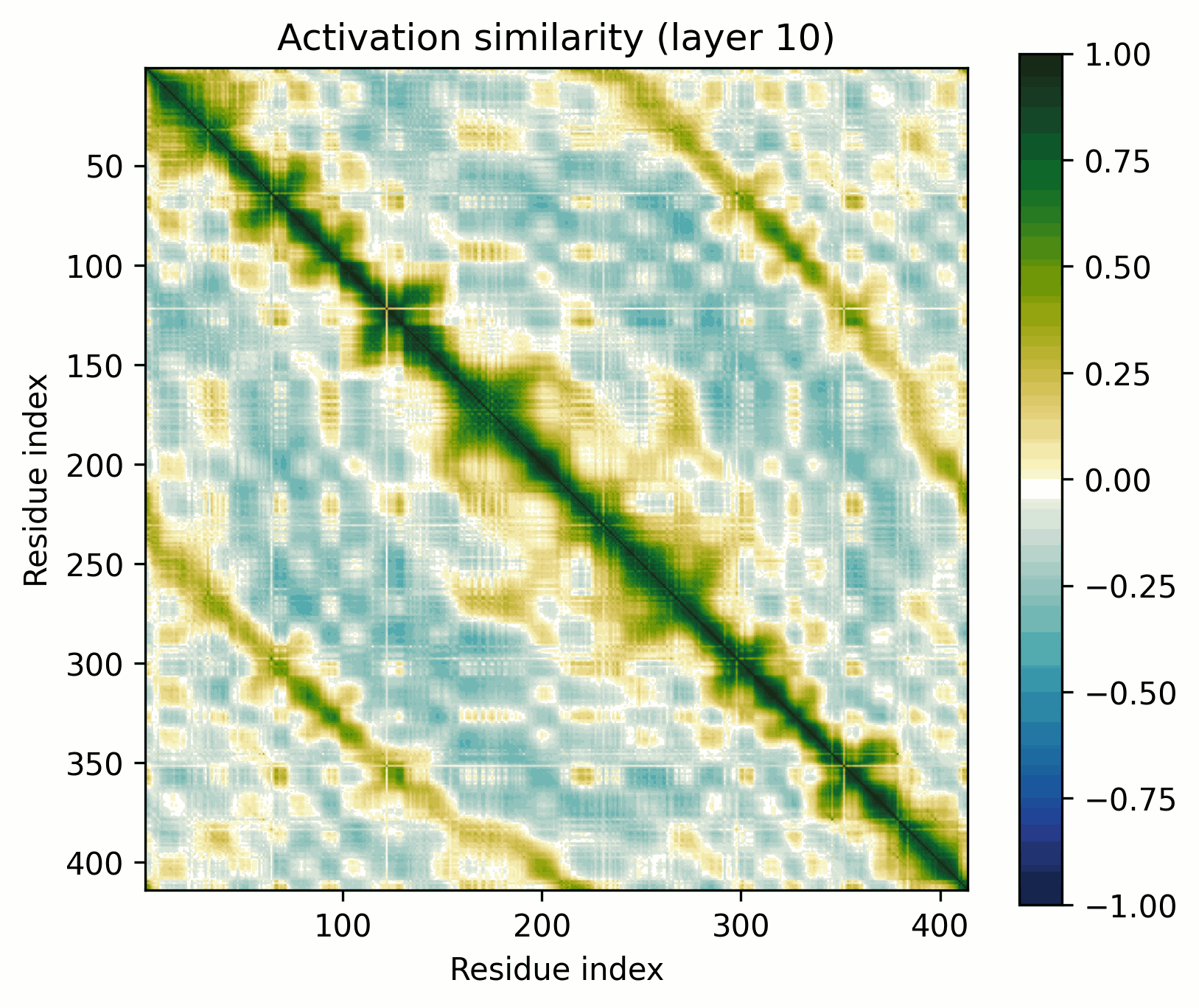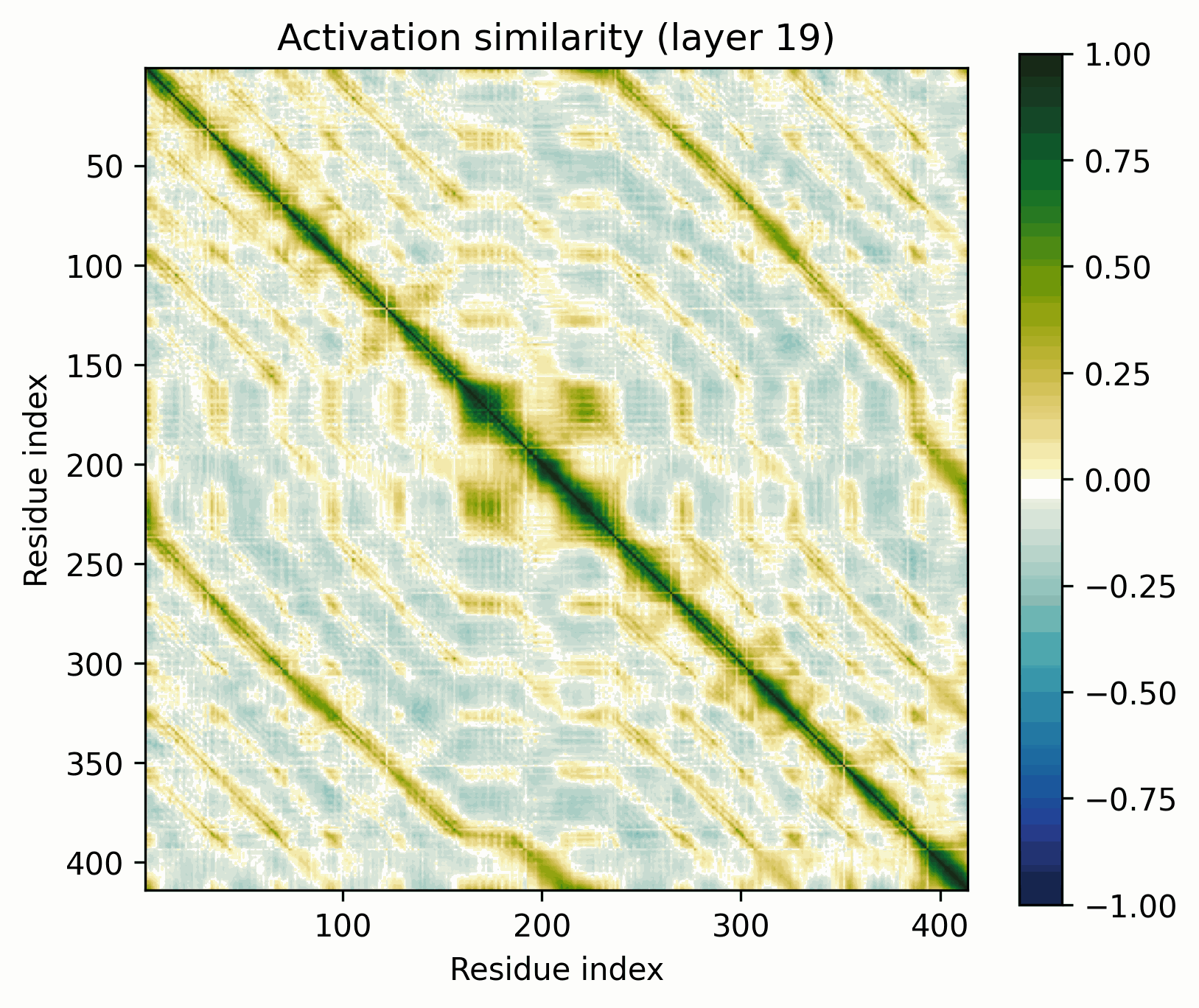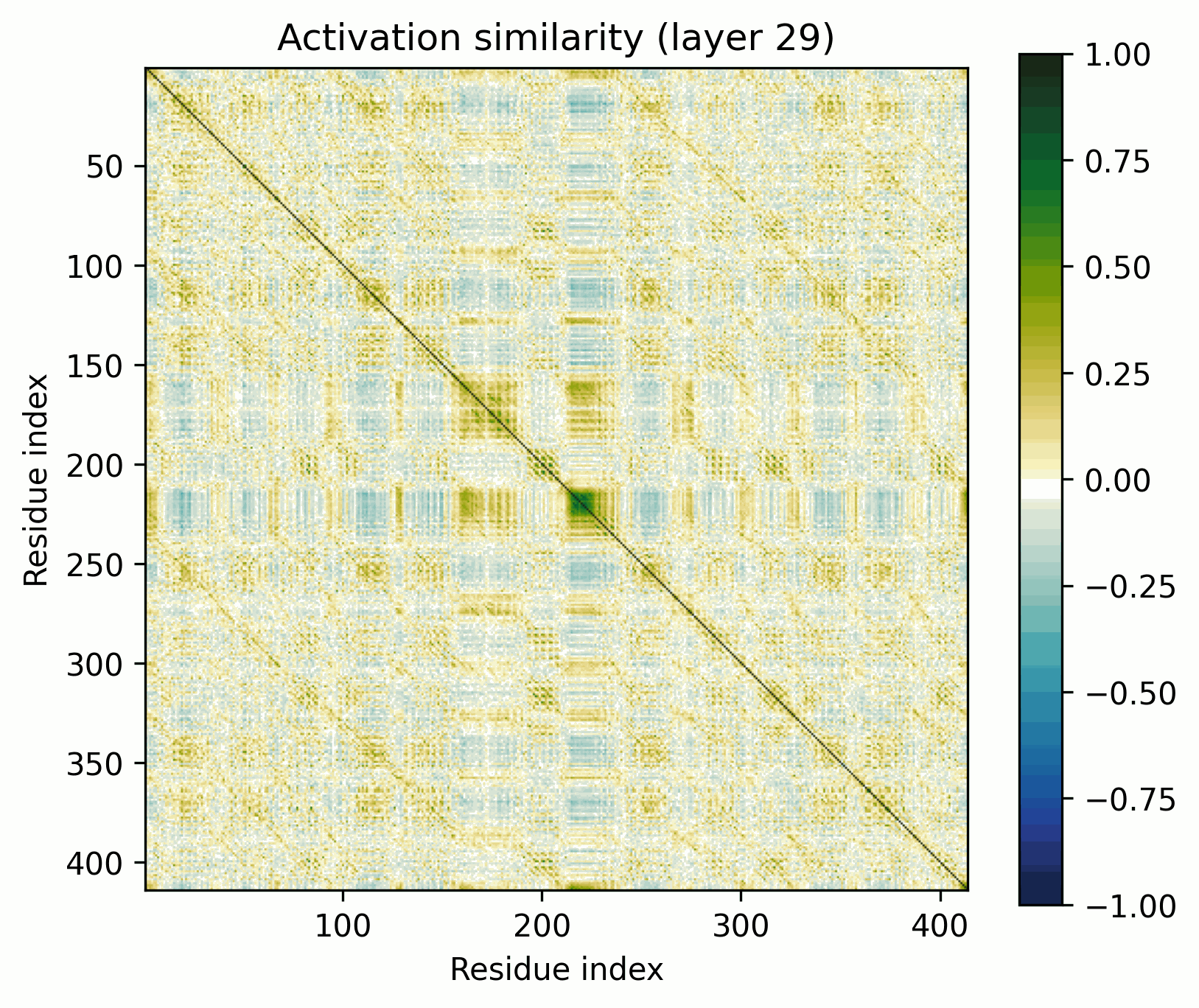
The pattern broadly resembles that of the alpha-barrel, but there's an obvious off-diagonal stripe.
As previously discussed, it’d be trivial to construct a model that would show this stripe at initialization if the two domains were position-by-position identical. That’s not exactly the case here, but this is still a fairly clear-cut instance of the ambiguities to which activation-space interpretability is susceptible. Has the model actually learned to represent the domain architectures per se, or is what we see a consequence of more trivial behavior? (My money is definitely on learned).
Beta Barrel
Let’s look at a beta barrel, E. coli outer membrane protein A. (Uniprot: P0A910, PDB: 1BXW)
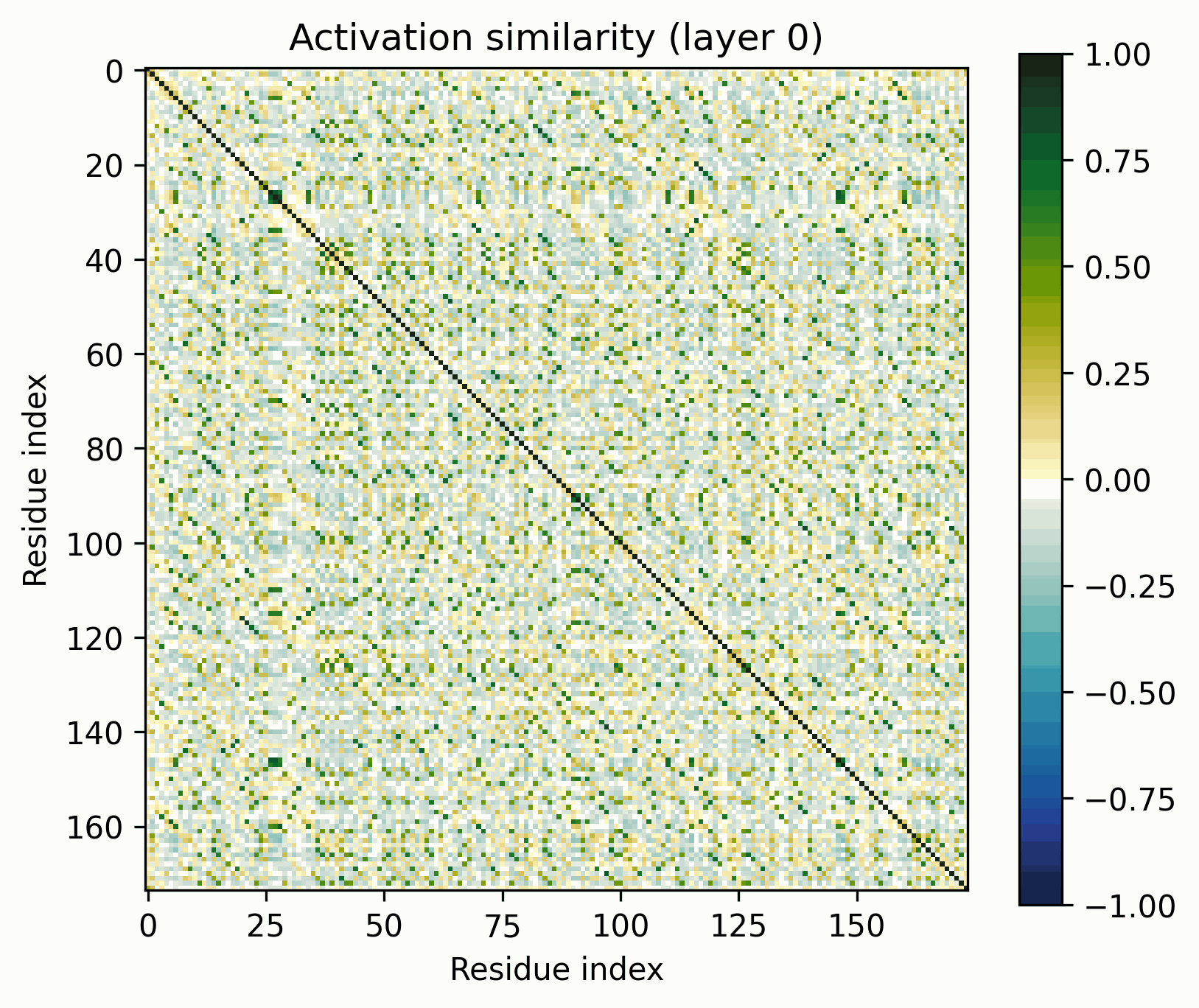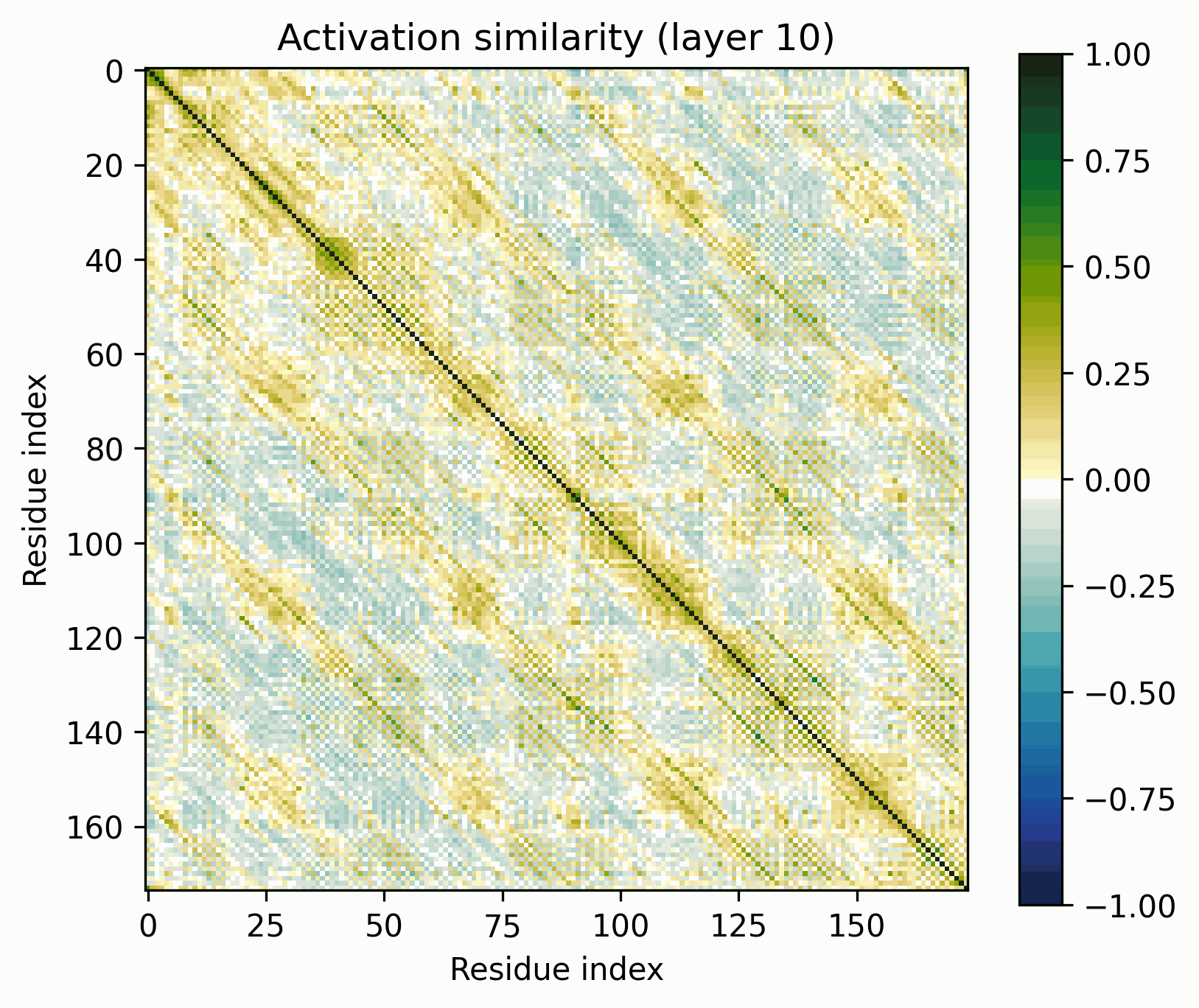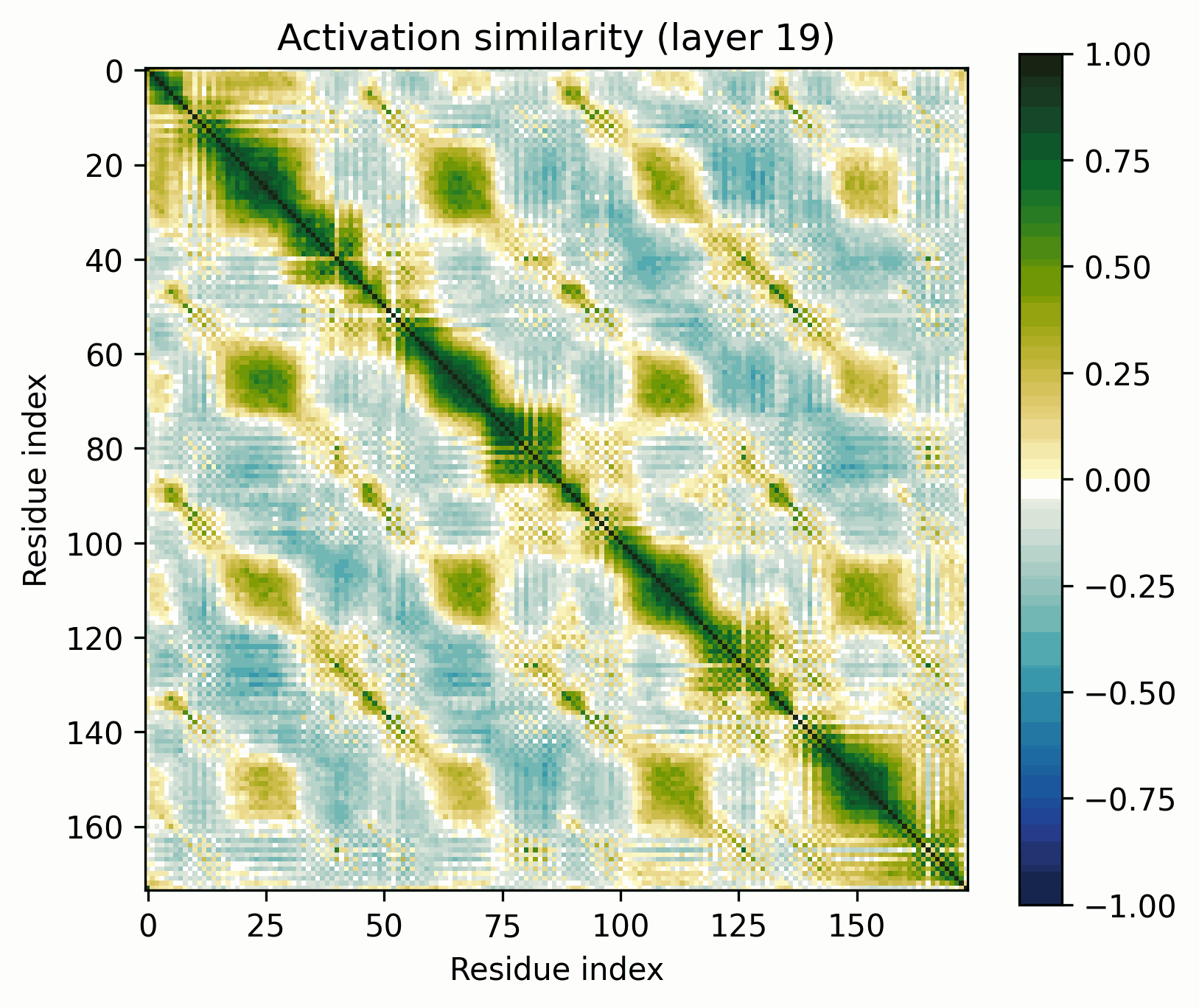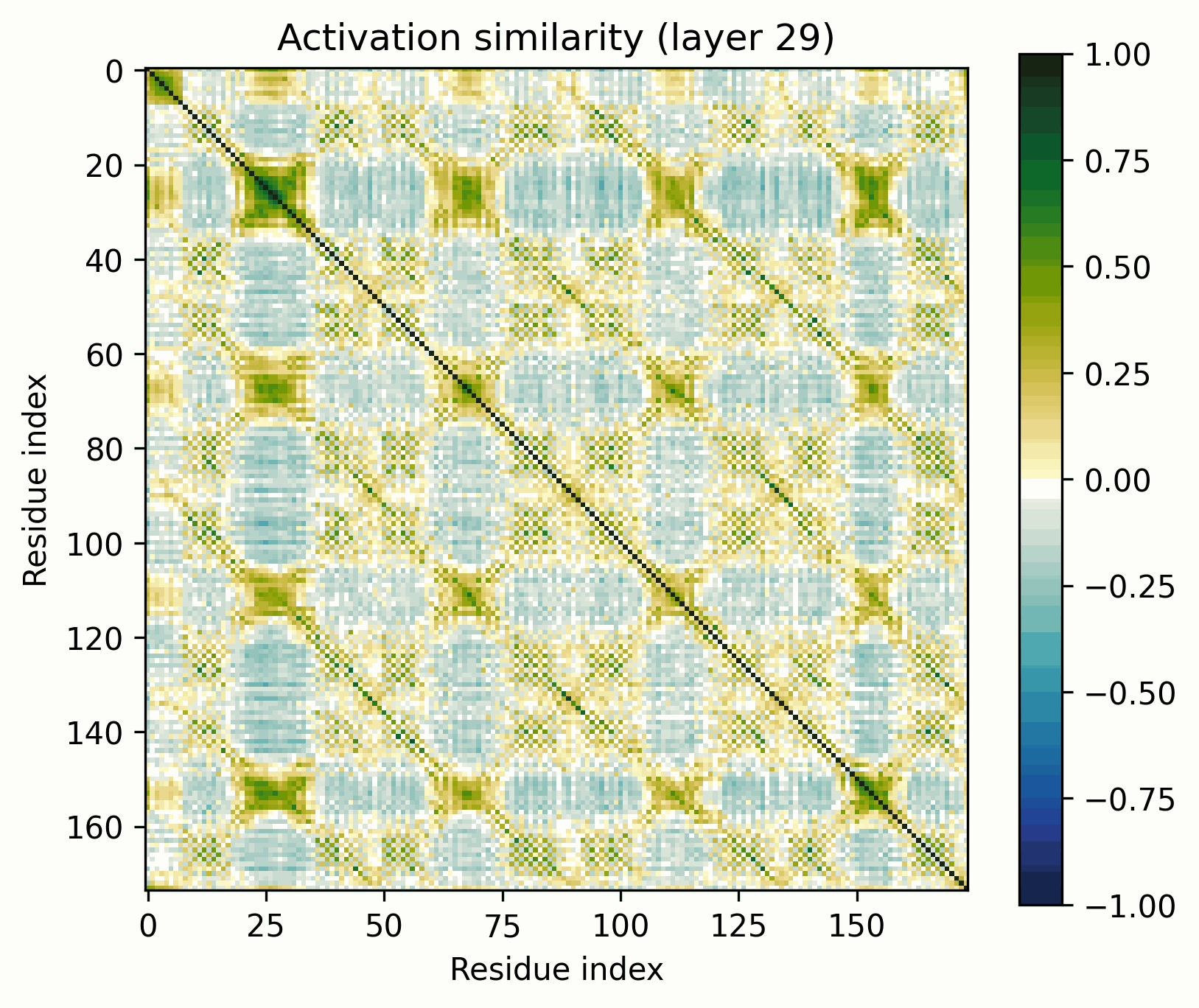
Observations:
- A clear pattern emerges in the later layers. Its representation divides cleanly into the β-strands that form the barrel wall and the connecting loop segments.
- The similarity of this organization to the alpha barrel representation is striking.
- The representation is often segmented into pairs of strands that share a short loop, particularly in the final layer.
Adenylate Kinase
The proteins we've inspected so far (besides PETase in the introduction) have repeating, homogenous structures. What do more generic globular proteins look like? Let's look at E. coli adenylate kinase (UniProt: P69441,PDB: 1AKE)[4].
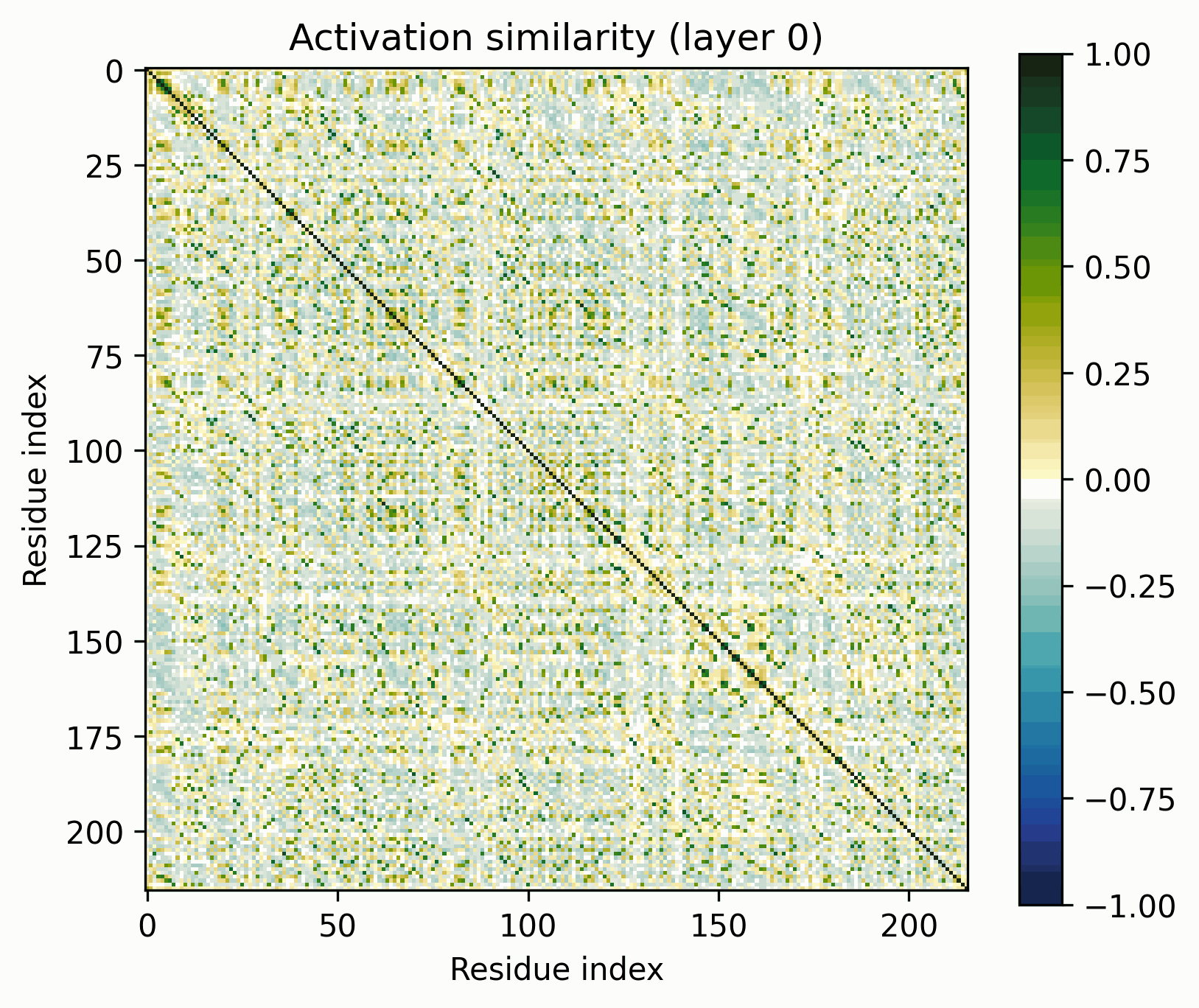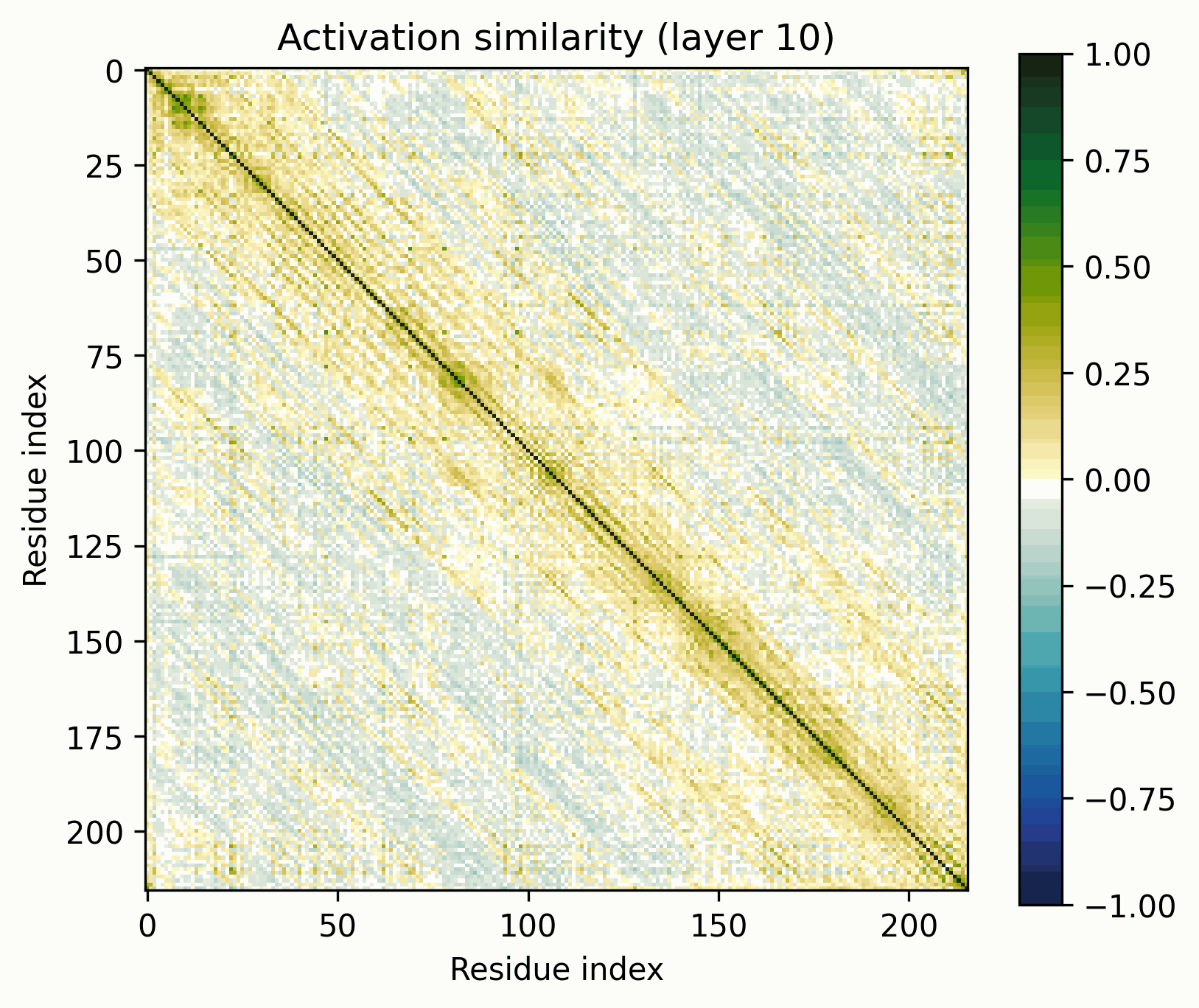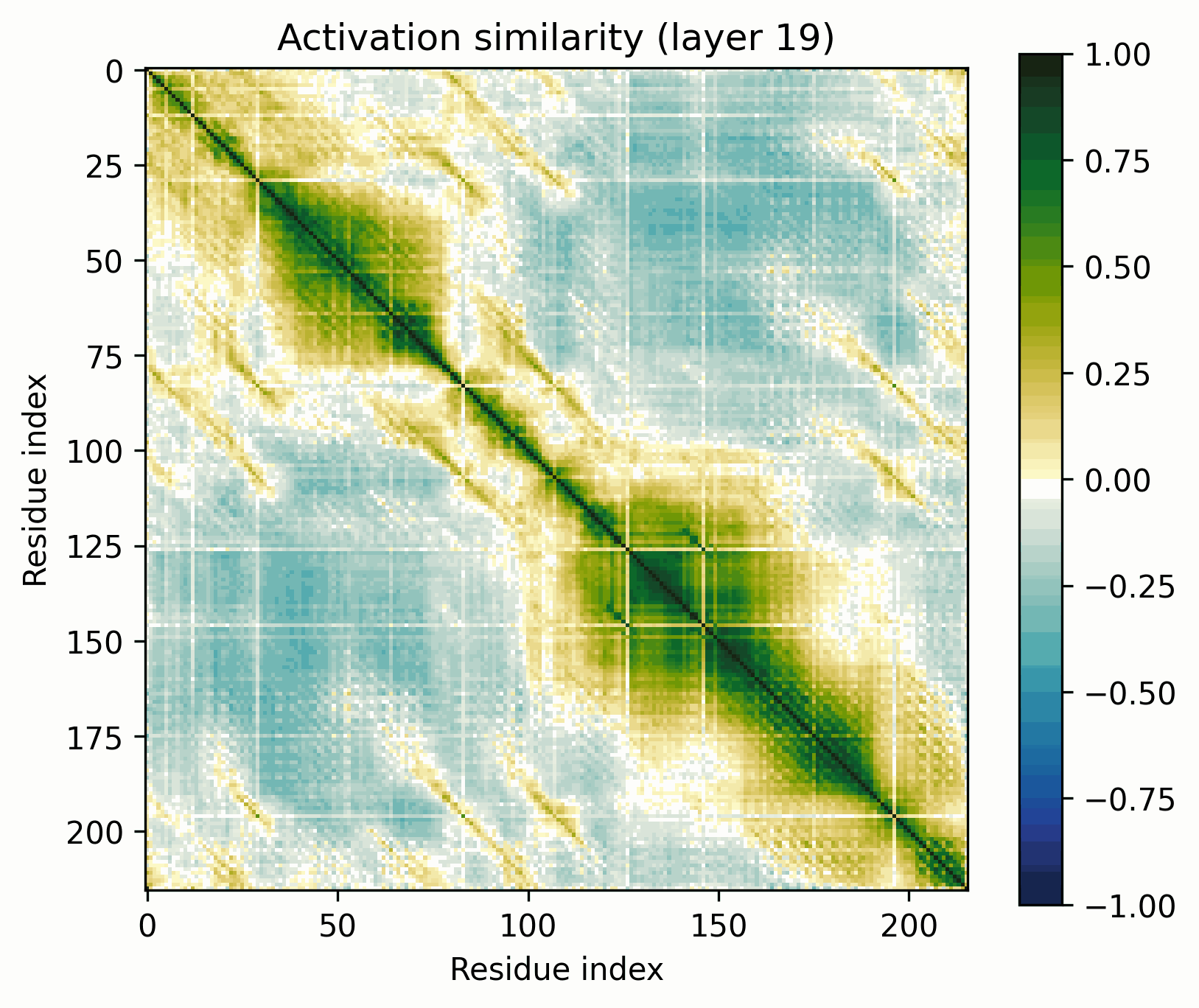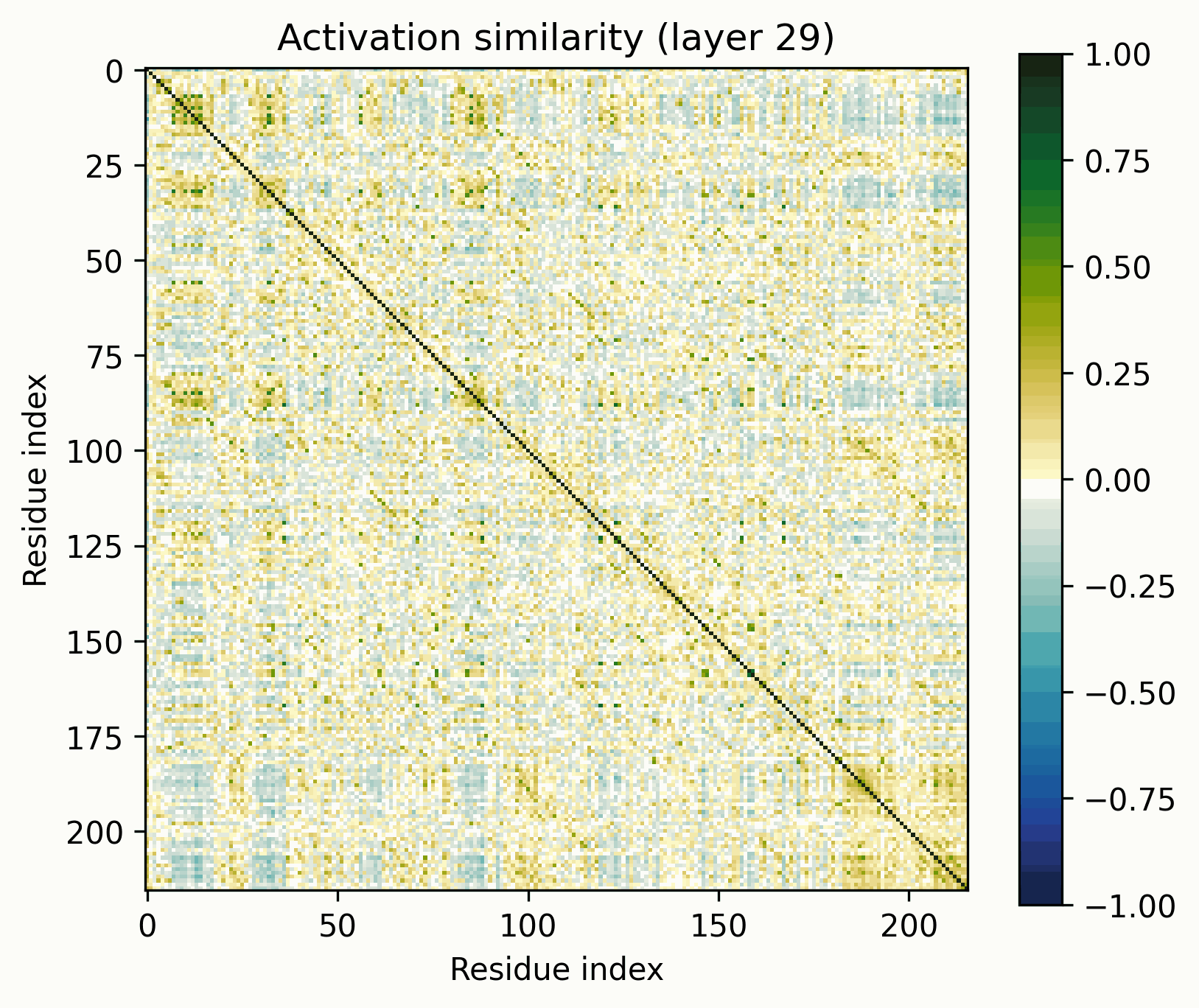
Observations:
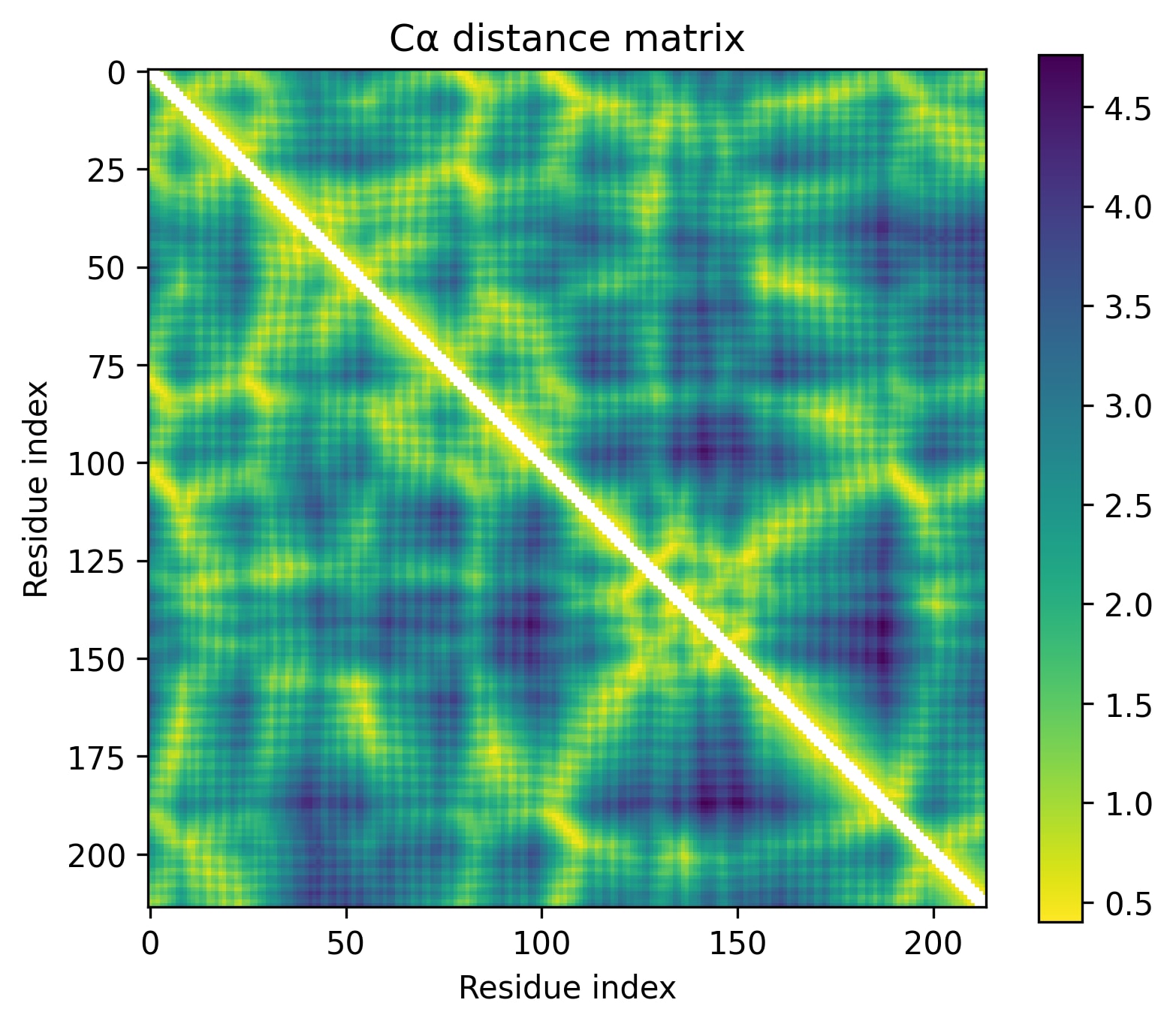
- Little has changed in the overall progression of the layers; the flow is qualitatively similar.Distinct positions that are orthogonal to all others and to each other stand out in the middle layers; these were rarer in the other examples.The protein is subdivided into a grid at several points in the progression; I am unsure what these correspond to. It's probably readily identifiable, but I want to avoid further scope creep, so it's left as an exercise for the reader.In the later layers, the most visually prominent features are a number of short diagonal stripes. A strong correspondence between these and the contact map is clear, though not a perfect one, and only for parallel contacts, which also manifest as diagonal stripes.
- Diagonal stripes going the other direction – top right to bottom left – are nearly nonexistent, despite several large ones being prominent in the distance map. Anecdotally, I do sometimes see these, but it's inconsistent.The largest two such diagonals span roughly 25-75 and 100-200. These regions are, at points, clearly segmented. Within each segment, similarity slowly decays off the diagonal.A tentative hypothesis: the primitives that correspond to these domains resemble positional embeddings more closely than the linear features found by SAEs.
Ablations
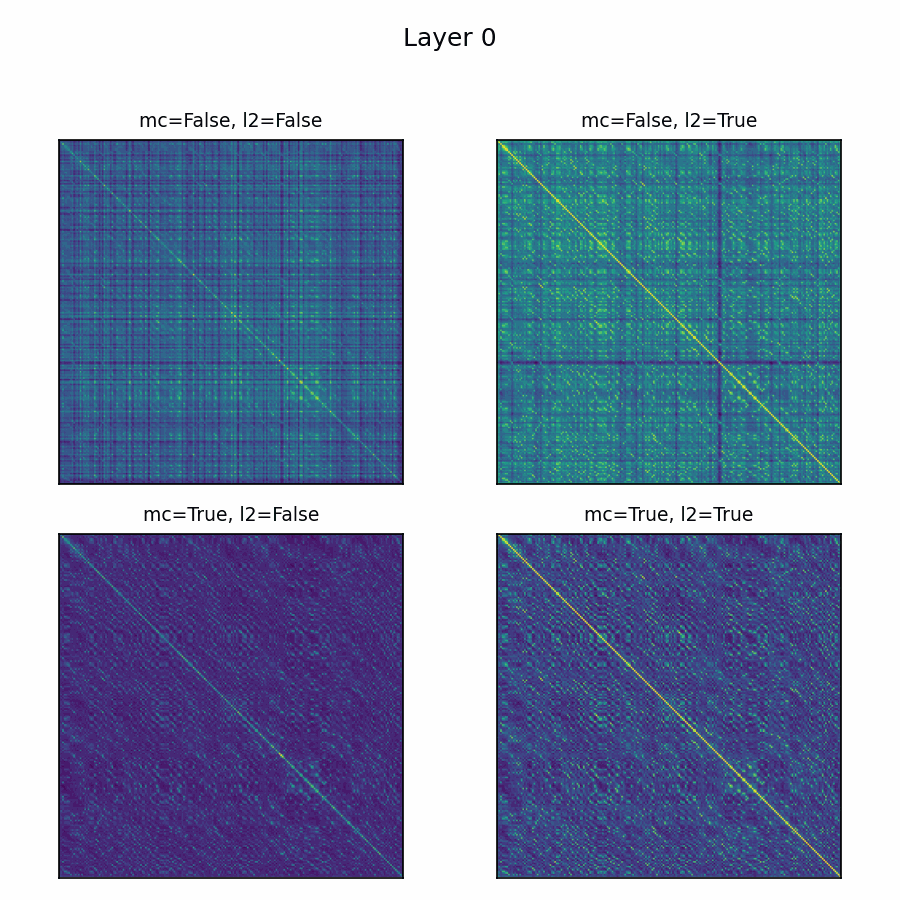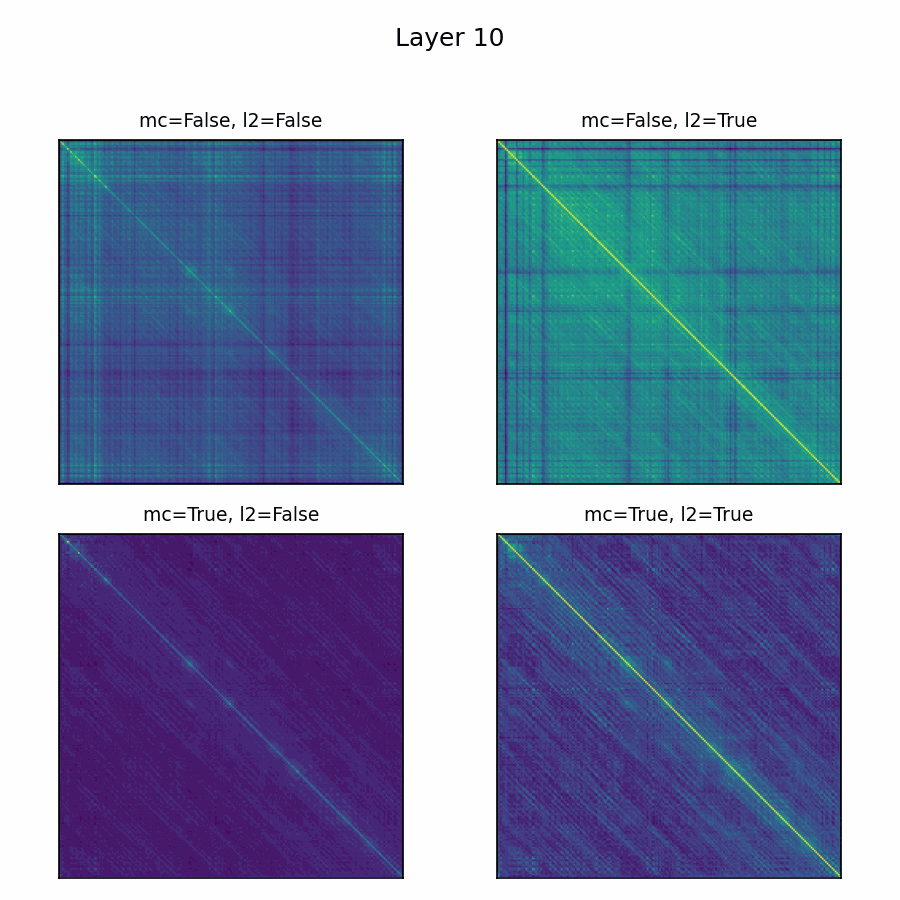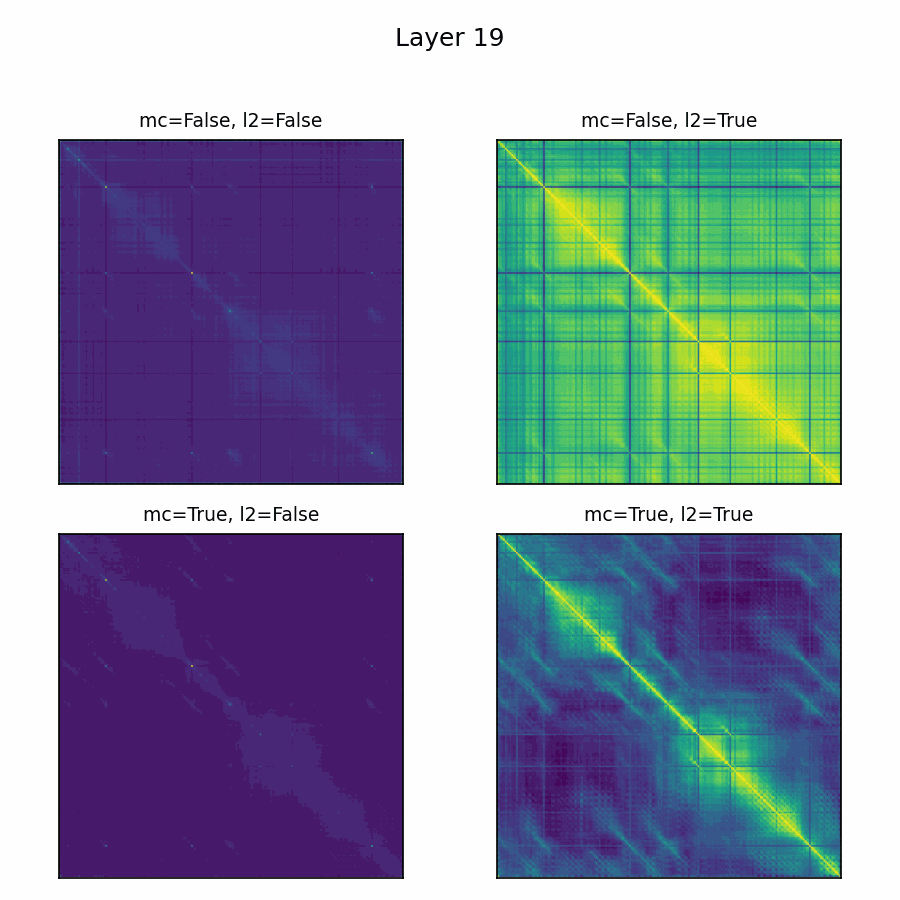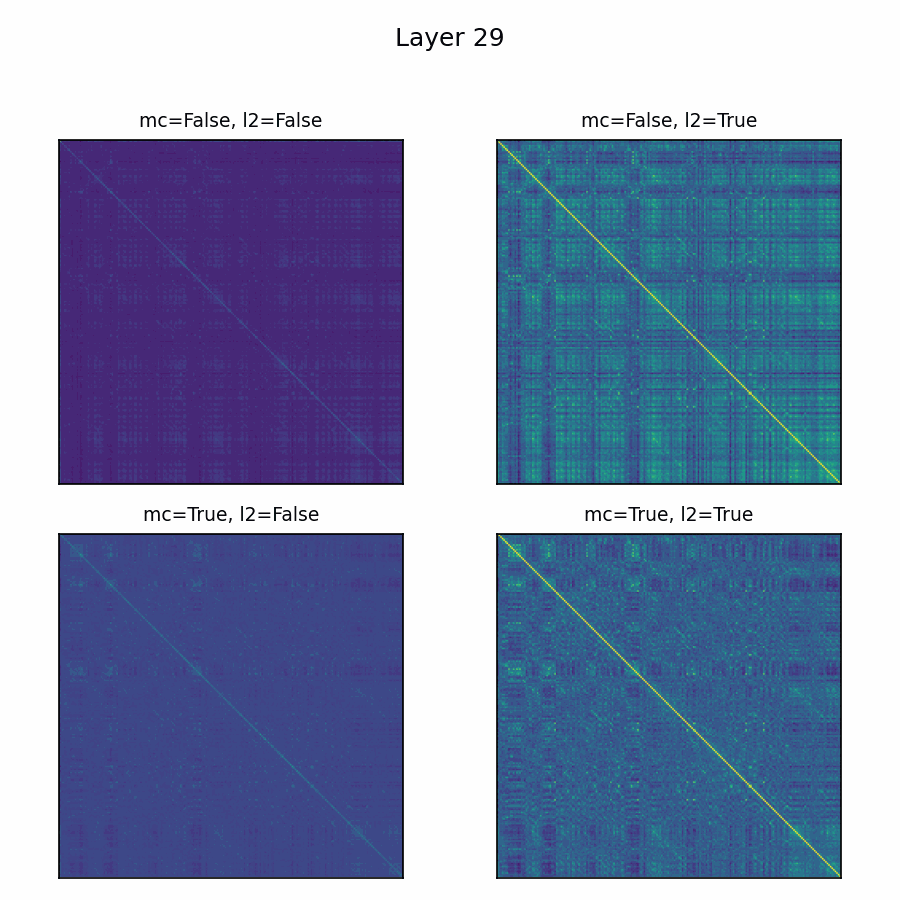
Here’s the adenylate kinase again showing similarities with and without mean-centering and normalization. Viridis is used here, with matplotlib's default scaling per plot and layer. Diverging colormaps performed terribly: with a strong offset, nothing's special about 0, so viridis provided the fairest shake. The plots without the normalization are far less illuminating.
Future Directions
This project has seen major scope creep, so I must limit the scope of what’s presented here. There are 30 layers and tens of thousands of protein families: I need to resist the temptation to add any more analysis and focus on the DNA language model. Of course, there is plenty more analysis to be done, including:
- Many proteins have a small number of amino acids ~orthogonal to every other position in the protein, including each other. Before normalization, they often have a very large magnitude. What are these tokens?How can the underlying features of a layer with extensive diagonals be captured, if not by SAE? Alternatively, how could a layer be structured to transform intermediate SAE-compatible latents to the diagonal-features, or sidestep the need for such features altogether?Is mean-centering and normalizing canonical somehow, or are there other transformations which work better?What might the Gram lens reveal in language models?
- ^
I don't think this distinction has been proposed before, if there already exists terminology for it, let me know and I'll emend this post.
- ^
This example and layer are slightly cherrypicked for aesthetics. All long-alpha-helices proteins I investigated had this pattern quite clearly, and across many layers, but there are often other motifs present. Any would have worked, so I opted to choose the one that makes this pattern as clear as possible.
- ^
An informal note: modeling alpha-helical barrels in this way, via a sine-like oscillation with a higher-frequency component at the crests – a grid on top of a grid – seems to be universal across biological sequence models. I’ve seen it in every model I’ve investigated, across architectures, trained on both raw DNA and proteins, with autoregressive, MLM, and other objectives. It just keeps showing up. I anticipate there are many more universal features like this.
I'm keeping this as a footnote because, while I strongly recall encountering it in my DNA language model, I don't have those data in front of me and don't want to have to eat crow if I cannot find it again or if it looks different from what I recall.
- ^
Chosen by asking o3 "Please choose a monomeric, soluble, globular E. coli protein, neither too long nor short, something nobody could begrudge if it were chosen as an exemplar to analyze the activations of an ESM model."
Discuss
