
随着地缘政治局势的不确定性、全球供应链的波动以及环境法规的日益严格,现代工业化学合成正面临前所未有的挑战。为响应可持续发展与循环经济的需求,化学品生产迫切需要转型升级。
2025年6月27日,韩国基础科学研究院与波兰科学院的研究团队在Nature Reviews Bioengineering上发表前瞻性观点文章,探讨了计算辅助合成规划(Computer-Aided Synthesis Planning, CASP)在绿色、可持续及循环化学领域的应用潜力。文章重点聚焦化学合成与生物工程的融合,以期推动更加环保、高效的化学品合成路径设计。

研究背景
计算机辅助有机合成研究最早可以追溯至20世纪60年代。近年来,随着人工智能和化学信息学的发展,合成算法已能够在逆合成与前向反应预测任务中规划出化学上合理且可经过实验验证的反应路径。
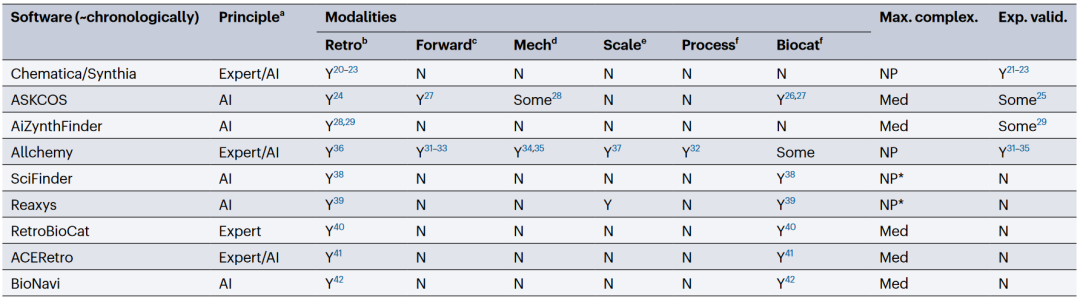
表1 现代合成规划软件工具示例
作者提议,在实现化学正确性之后,CASP的下一步应着眼于“绿色性、可持续性与循环性”三大核心目标,辅助科学家规划环境友好、资源节约、废物可回收的合成方案。这一目标的实现离不开化学家与生物工程师之间的紧密协作——例如共建绿色指标体系、发展面向酶催化反应的智能算法等。
作者设想,未来化学合成可与AI和生物工程深度融合,为制药等化学品生产带来深远变革,减少对化石原料的依赖,推动产业可持续发展。
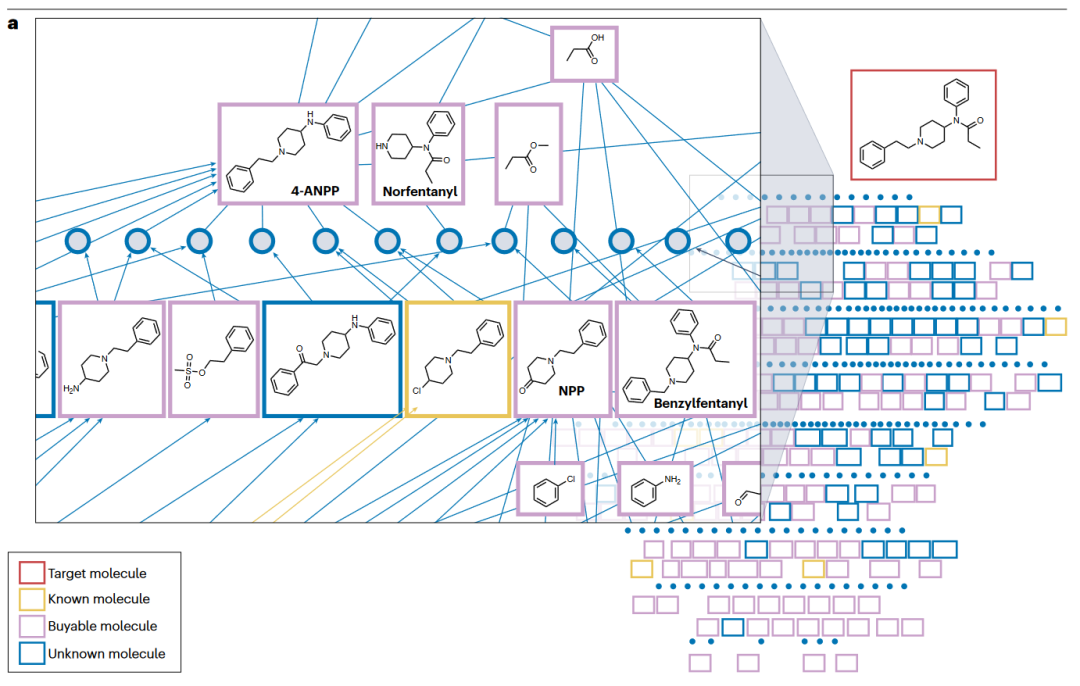
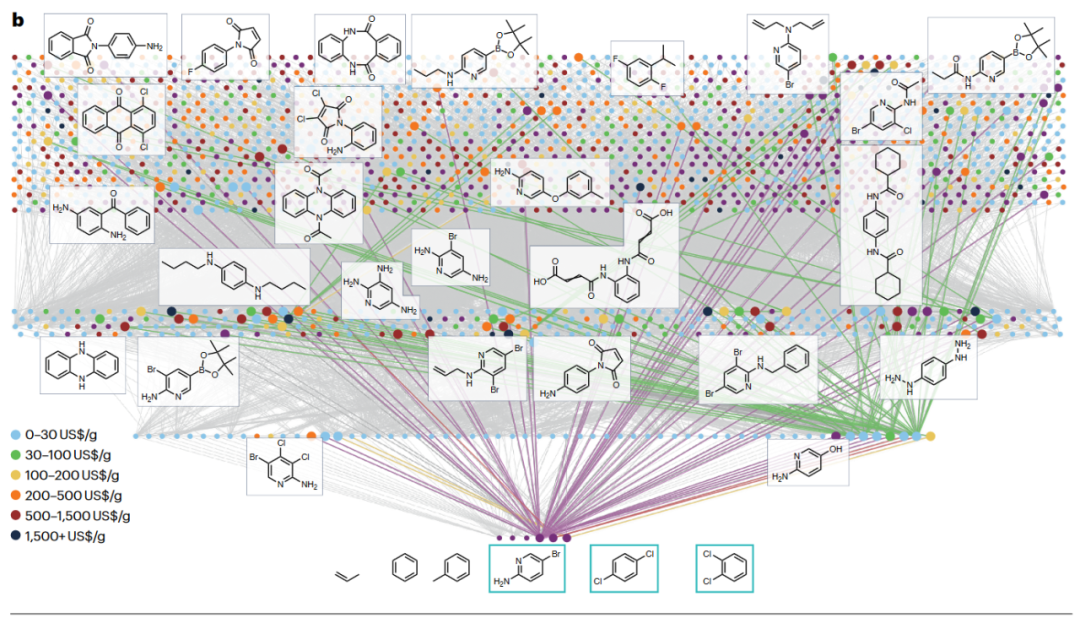
图1 前向和后向计算机辅助合成
绿色、可持续、循环化学的核心概念
三者虽目标一致,均致力于减轻化学生产对环境的负面影响,但在路径与侧重点上有所差异:
绿色化学(Green Chemistry):
目标:减少化学污染,最小化或消除有害副产物;
特征:强调原子经济性、能效优化、安全溶剂、可再生原料、催化和生物催化等;
适用于线性"取-制-用-弃"型生产模式。
可持续化学(Sustainable Chemistry):
定义:寻求提高自然资源利用效率以满足人类对化学产品和服务需求;
重点:维持和延续生态健全的发展;
与绿色化学的区别:关注"环境风险最小化"vs"生态健全发展的维持"。
循环化学(Circular Chemistry):
目标:用循环模式替代线性经济模式,实现材料在价值链中持续循环再利用;
理念:承认废物是化学生产的固有产物,将其视为有价值的起始材料;
促进:废物再利用、资源保护、原子循环、能源和工艺效率。
对于算法的预期是能设计强大的合成路线,可以最大程度地减少反复试验。对于满足这一要求可以有以下几种考量。
绿色性:规划的路线可以绕过有害化学物质,促进催化和生物催化方法,并可以优先考虑原子经济。
可持续性:优选能效高、以可再生资源为原料的路径。
循环性:可以以“前向合成”模式循环运行,即重新利用其他过程中的废物,且理想情况下让功能性分子易于生物降解。
此外,还需引入满足生命周期分析的多因素指标,以评估其对资源,生态系统和人类健康的潜在影响。
计算机辅助化学合成中的实践应用
从废物出发的药物合成路径设计
文章展示了一项从189种工业化学废物出发通过简洁和环境友好的路径将其转化为重要药物的算法研究。具体流程如下:
网络生成:从废物分子集合和常见试剂出发,使用数万个反应规则进行正向传播。
产物识别:在数亿个假定产物中筛选出近200种FDA批准的药物和及百余种工业农用化学品。
路径回溯与排序:建立从产物到底物的合成路线,依据绿色化指标进行优先级评估
其中评分标准涵盖试剂毒性,极端反应条件,非收敛性合成等因素。实验验证显示,算法设计的多条合成路径在良性条件下能有效进行,此外还能合成如卡维地洛、比索洛尔、丙氧他卡因等药物。
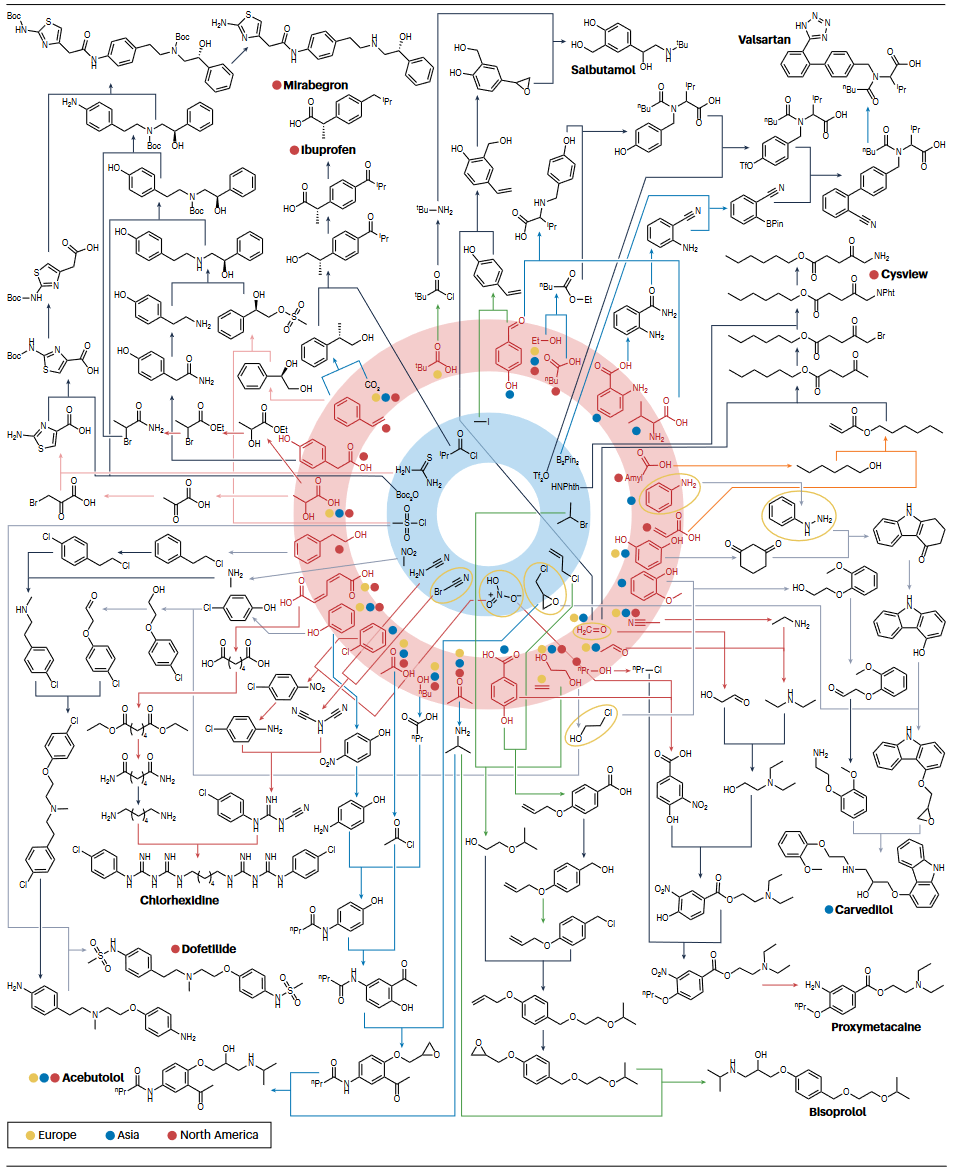
图2 从废物底物来源的药物算法设计合成网络片段
降解合成与循环性潜力挖掘
可控降解可以增加用于真正循环合成的废物衍生池以及潜在可增值的底物池。在传统合成化学中,研究人员通常较为熟悉诸如水解、脱羧等基本的降解反应,而对复杂降解过程的应用则相对有限。然而,对于计算机而言,只要掌握了足够的反应规则与机制知识,即可有效识别并实现更复杂的结构转化。具体来说,合成规划算法通过学习近千种降解反应类型和类似数量的辅助基团转换或重排反应,从而在结构拆解与重构中更深入地理解反应规律,并据此设计出具有潜在增值价值的合成路径。
以降解相对便宜的奎宁(约4-5美元/克)为例,算法通过7轮网络传播产生近7000个分子,最终找到29个商业可得且价格至少高30倍(>150美元/克)的分子。且在绿色条件下经由实验验证,效率大大高于先前文献报道。
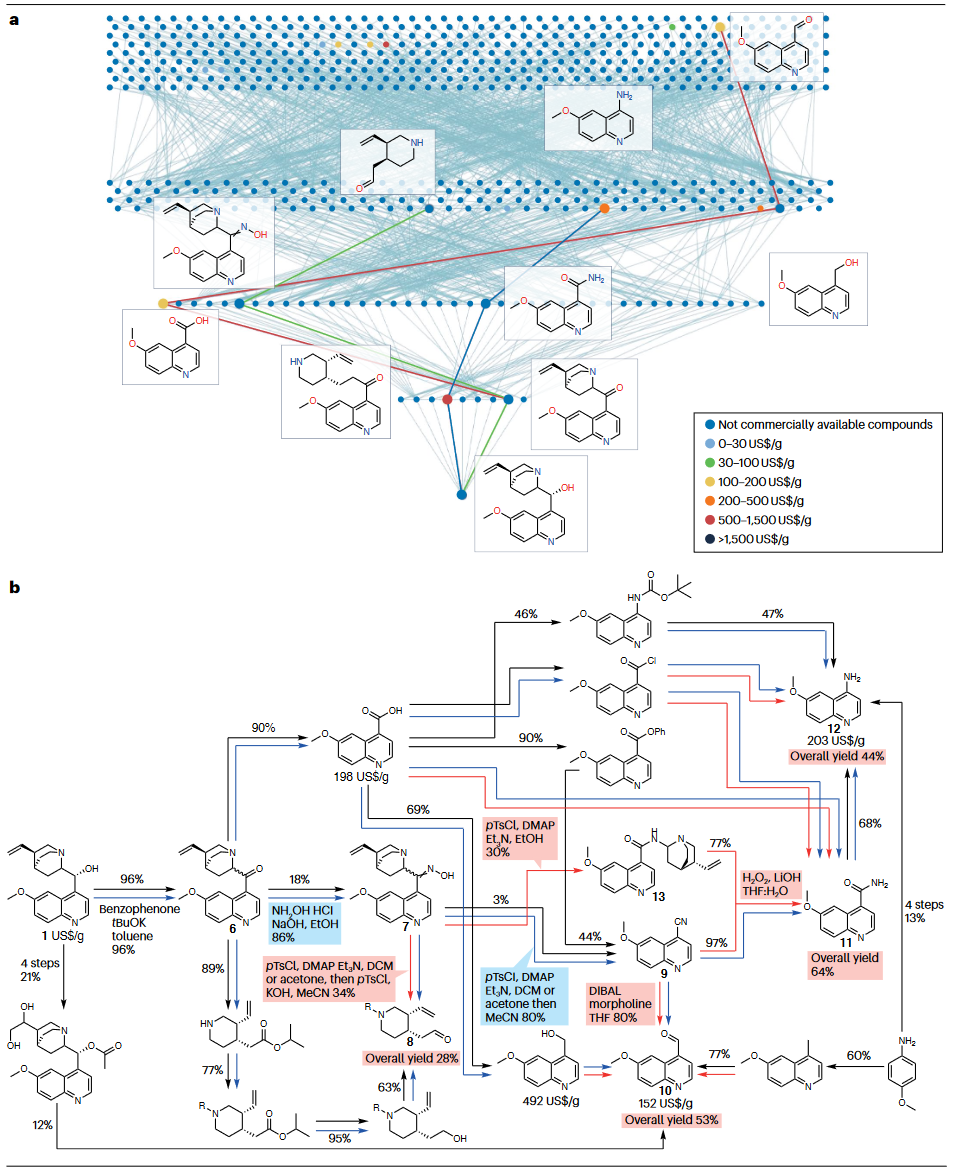
图3 奎宁降解为较小且增值化学品
化学合成与酶催化的融合挑战与机遇
酶底物范围预测的局限
当前酶反应预测主要基于模板匹配,存在如下问题:(1) 数据偏差,数据库以成功反应为主,忽略失败因素;(2) 模板缺陷,忽视反应机制,难以判断底物是否适配酶的活性位点;(3) 数据规模不足,酶反应数据远少于有机反应(几十万 vs 数千万)。
人工智能在酶底物范围预测中的进展
当模型面对训练集中未见过的分子(以及较小程度上的酶)时,准确率大幅下降。这对化学合成中的应用至关重要,因为试剂和催化剂需要在最大程度上广泛且通常前所未有的底物集合上工作。
酶底物范围的预测可能不是人工智能问题,而是数据问题,需要更大、更多样化的数据集(包括正面和负面例子)来训练可泛化的模型。
解决策略包括:(1) 鼓励实验失败数据的公开,建设开放数据库;(2) 借助自动化反应平台生成大规模数据;(3) 注重底物多样性、商业可得性及结构空间覆盖度。
未来,这些模型可嵌入CASP工具,对每个逆合成候选步骤进行酶实现可能性的评估,实现酶催化与合成规划的无缝集成。
展望
尽管当前仍面临算法与数据的诸多挑战,计算机在合成科学中的作用已经并将持续深远,推动先进化学品的设计方式发生根本变革。回顾数十年前计算辅助合成(CAS)的初期探索,当时受限于计算资源、算法能力与知识库的匮乏,难以实现真正实用的合成路径规划。
而如今,随着算力的飞跃与算法的成熟,所有关键要素已基本就位,CAS 正在逐步渗透至合成设计的各个阶段:(1) 快速生成多条(从数十至数百)合理的合成方案;(2) 筛选出可扩展的、具备工艺可行性的路径;(3) 引入经济性、工艺性与绿色化等多维评分体系,进行多参数优化。
这种多参数并行评估能力是计算系统相较人类专家的核心优势。传统上,化学家往往采用线性流程进行合成设计,例如先设计路线,再查询底物价格,最后分别优化其他参数。而算法则可整合多源知识,针对多个目标同步开展优化,使合成路径更加智能、高效和全面。
尽管基于化学的合成设计工具已取得长足进展,未来真正具有突破潜力的研究机会存在于化学与生物工程的交叉地带,主要体现在以下两个方向:
(1) 绿色性指标的拓展与评估。引入毒性、环境影响等生物相关性指标,用于更全面地评估合成路径的绿色程度和生命周期可持续性。
(2) 酶催化模型的开发与主流化。酶作为理想的催化工具,在工业与学术界尚未广泛应用,其潜力有待释放。尤其是酶的底物范围预测,将成为生物合成能否大规模集成进CAS系统的关键所在。
尽管当前普遍寄希望于机器学习方法在酶范围预测方面的进步,但作者指出,数据质量和多样性才是影响预测泛化能力的决定因素。作者提出两条新的研究路径:
(1) 探索更优的分子表示方式能更好表达三维结构与原子间相互作用的表示方法。
(2) 重审经典模拟方法的新可能。重新评估如分子动力学模拟与分子对接等传统方法在当前计算力背景下的潜力,尤其是在高通量、系统性使用条件下。
作者提议不拘泥于当下AI的流行风潮,采用多元策略与工具,构建更准确、更具泛化能力的酶作用范围预测系统,进而补强CAS工具链,拓展其在绿色、可持续合成中的实际应用边界。
参考链接:
https://doi.org/10.1038/s44222-025-00312-7
--------- End --------
内容中包含的图片若涉及版权问题,请及时与我们联系删除
