
DRUGAI
当前蛋白质组基因组学在解析基因表达复杂性方面面临重大挑战。为此,研究人员开发了 moPepGen——一种基于图结构的算法,能够在保持线性计算复杂度的同时,全面生成非典型肽段(non-canonical peptides)。moPepGen 可灵活适用于多种测序和质谱技术,覆盖不同物种,并支持对各种基因组和转录组变异的解析。在人类癌症样本中,moPepGen 成功揭示了大量先前未被观测到的非典型肽段,这些肽段来源于种系与体细胞突变、非编码开放阅读框(ORF)、RNA 融合与环化等事件,展示了其强大的跨平台、跨物种、多模态整合能力。

一段 DNA 序列可以通过多种机制生成多样化的蛋白质产物。这些机制包括基因组变异(如点突变、插入缺失)、转录过程中的可变剪接、RNA 编辑、RNA 融合以及翻译后修饰等。RNA 环化更进一步扩展了编码潜力,使非线性模板也参与蛋白翻译过程。由于上述层级组合方式呈指数增长,蛋白质组学技术即便快速发展,仍然难以覆盖全部蛋白形式,尤其是那些不属于标准蛋白数据库的非典型肽段。
当前检测这类肽段的常用方法包括从头序列推断(de novo sequencing)与开放式搜索策略。然而,这些方法往往计算代价高、误报率或漏检率居高不下,数据解释和变异定位也较为困难。因此,主流蛋白质组研究转向构建样本特异性数据库,以结合 DNA 和 RNA 层面的变异信息来缩小搜索空间、提高检测灵敏度。
然而,构建这类数据库的关键在于是否能准确模拟转录、翻译和肽段剪切的复杂过程,并穷尽性地生成潜在蛋白产物。已有方法多数仅处理某一类突变或某种类型的剪接事件,导致假阴性比例升高,甚至在数据库缺失目标肽段时产生误检。为克服上述限制,研究人员提出了 moPepGen 框架,借助多层图结构模型,系统性整合所有变异组合,并生成更全面的非典型肽段数据集。
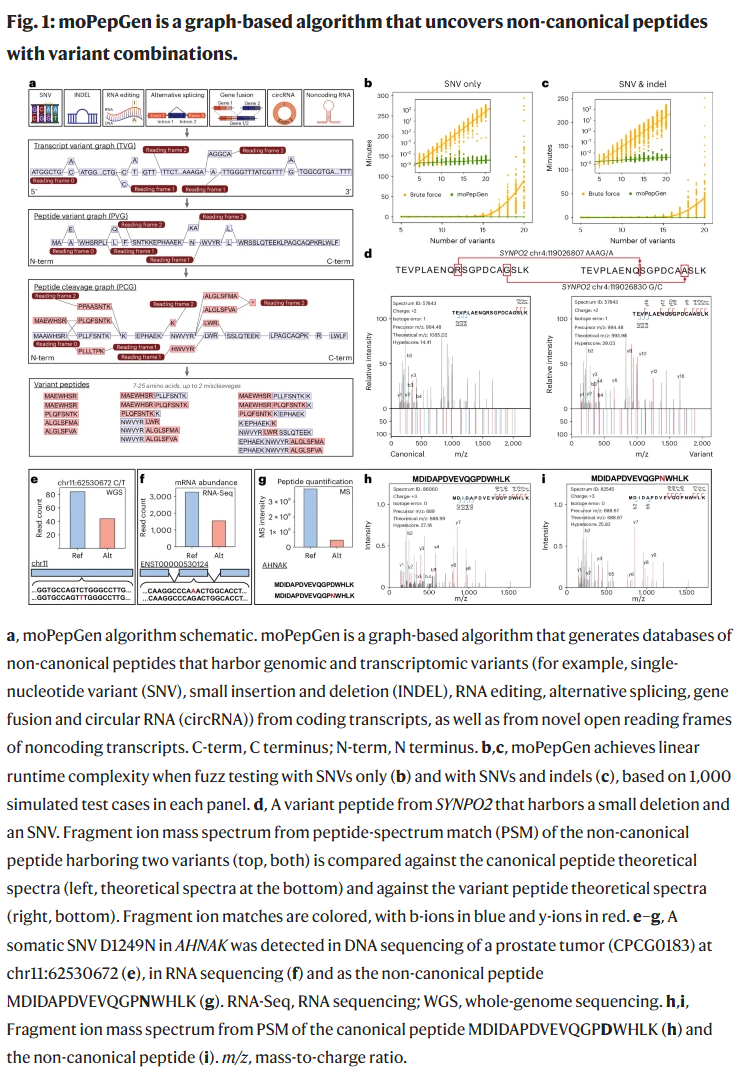
算法原理与性能验证
moPepGen 将每个转录本构建为三种互补图结构:转录本变异图(TVG)、肽段变异图(PVG)和肽段剪切图(PCG)。通过整合各种小变异(如 SNP、indel、RNA 编辑位点),以及非典型转录本骨架(如新ORF、融合转录本、RNA环化等),该框架可系统性地遍历所有可能的变异组合,并模拟肽段的翻译与酶切。
研究人员首先通过百万次模糊测试验证 moPepGen 在模拟环境下的准确性,结果显示其输出与理论穷举算法完全一致,同时具备线性时间复杂度。
与现有工具对比分析
研究人员进一步将 moPepGen 与现有两个自定义数据库生成工具 customProDB 和 pyQUILTS 进行对比,使用五例前列腺癌样本的多组学数据进行评估。在处理基础突变(如种系与体细胞点突变、indel)时,三者性能相近,但 moPepGen 在复杂生物学场景(如可变剪接、RNA编辑、环化、融合等)下是唯一能全面建模所有类型变异的工具,准确率与敏感性明显优于其他方法。值得注意的是,超过 80% 的非典型肽段为 moPepGen 独立预测,仅有 3% 未被其识别,这部分由其他工具所作生物假设所致。
多样本应用验证
研究人员在多个真实样本中展示了 moPepGen 的实用性:
酶切多样性分析:在人类扁桃体深分级样本中,使用七种不同蛋白酶处理,moPepGen 成功检测到 1,787 个来自非编码转录本的新 ORF 肽段,其中 Arg-C 表现最佳,显示不同酶切方案可辅助提升检测覆盖率。
小鼠蛋白质组应用:在小鼠 C57BL/6N 模型中,moPepGen 基于种系变异信息生成并识别了共计超过 20,000 个非典型肽段(包括编码区与非编码区 ORF),其中在脑、肝、子宫组织中共检测到 343 个非编码 ORF 肽段,证明其跨物种适应性。
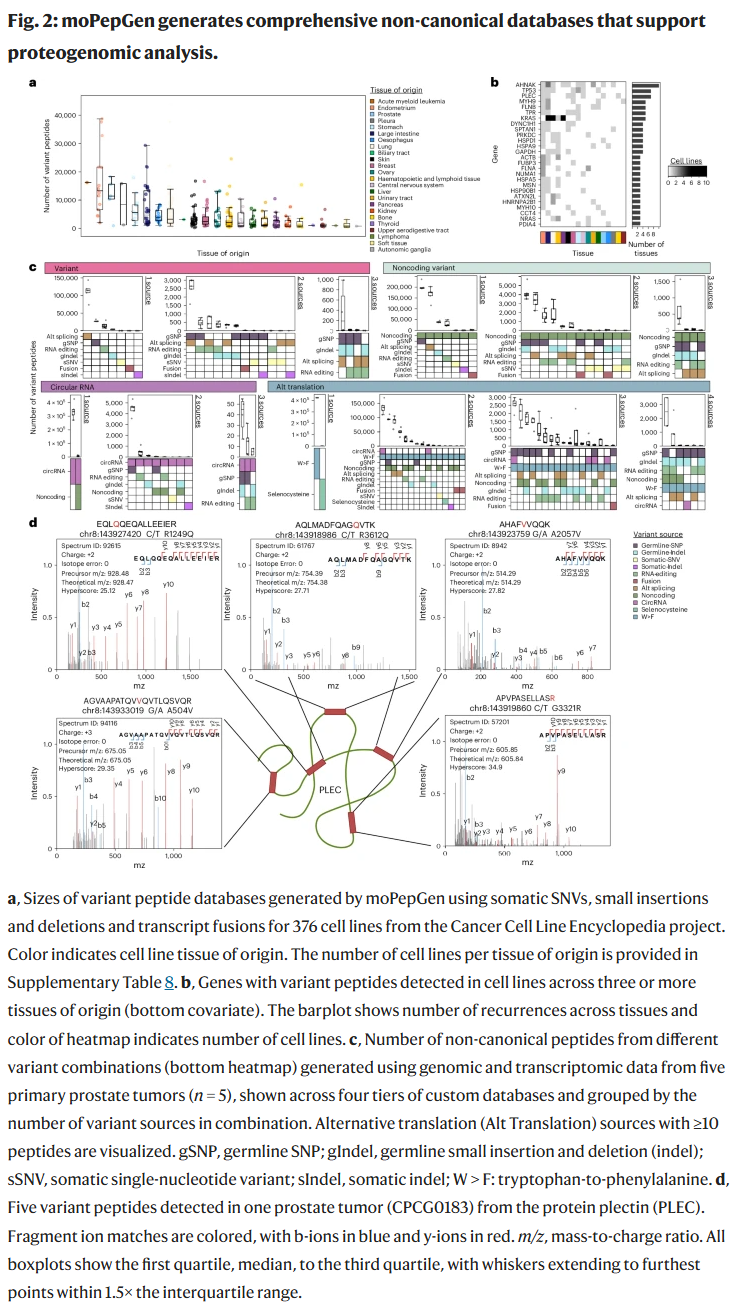
癌症细胞系:在 375 个人类癌症细胞系中,moPepGen 平均为每个样本生成 2,683 个候选肽段,并检测到平均 39 个,涉及 TP53、KRAS、HRAS 等关键癌基因。部分肽段可作为潜在新抗原,在多细胞系中重复出现,暗示其功能重要性。
数据非依赖采集(DIA)质谱:在透明细胞肾癌样本中,研究人员检测到超过 300 个变异肽段,主要来源为种系 SNP 和可变剪接,RNA 编辑事件也占一定比例,验证了该方法在 DIA 数据中的可行性。
前列腺癌样本分析:在五例深度测序的前列腺癌样本中,moPepGen 生成了超过 130 万个独特肽段,涵盖 115 种组合类别,检测到超过 200 个非典型肽段。其中一些肽段源自 KLK3(前列腺特异抗原)中的双突变区域,部分肽段由 circRNA 导致的反向剪接生成,在多个肿瘤样本中复现,说明其潜在的生物学意义与通用性。
讨论
moPepGen 提供了一种高效、全面的非典型肽段生成与识别方案,突破了当前蛋白质组基因组学在突变整合和数据覆盖方面的瓶颈。其核心优势包括:
高通量与可扩展性:可在常规计算资源上线性扩展至全基因组级别。
变异整合能力强:覆盖基因组与转录组多种类型变异,包括复合型变异与非编码来源。
跨平台适应性广:可适配不同的质谱平台、酶切方案和物种背景。
算法可解释性强:输出肽段均附带来源注释,便于溯源和交叉验证。
综上所述,moPepGen 作为一项新兴工具,能为变异肽段检测、癌症新抗原识别、非编码蛋白翻译研究等提供有力支持,有望广泛应用于基础研究、肿瘤免疫治疗靶点发现、以及大规模多组学数据库构建中。
整理 | WJM
参考资料
Zhu, C., Liu, L.Y., Ha, A. et al. Identification of non-canonical peptides with moPepGen. Nat Biotechnol (2025).
https://doi.org/10.1038/s41587-025-02701-0
内容中包含的图片若涉及版权问题,请及时与我们联系删除
