

Stanford CS224W: Machine Learning with Graphs
https://colab.research.google.com/drive/1TCRNimtb2c9_90nSEAEa2G-37OKVOAm7
背景
在这个项目中,我们专注于如何使用图神经网络来分析蛋白质共表达网络以协助疾病诊断。
蛋白质共表达网络是了解不同疾病背后的生物学机制和细胞类型变化的一种越来越流行的工具。这些网络是通过测量血液和脑脊液中数百甚至数千种蛋白质的蛋白质水平来构建的。然后可以根据邻接矩阵构建图,以反映蛋白质表达水平的相似性。这些共表达网络增加了蛋白质-蛋白质相互作用网络(测量蛋白质之间的物理相互作用)提供的信息,因为它们允许研究人员研究蛋白质水平在动态环境中如何变化。
在本教程中,我们使用来自阿尔茨海默病神经影像学计划 (ADNI) 的数据,重点介绍阿尔茨海默病(AD)。AD是最常见的痴呆形式,也是 65 岁及以上成年人死亡的主要原因之一。它的特征是极度记忆丧失和认知能力下降,在大脑内,它的特点是淀粉样斑块(淀粉样蛋白-ß 蛋白的细胞外沉积物)和神经原纤维缠结(由过度磷酸化的 tau 蛋白聚集体组成)和灰质丢失。近年来,蛋白质共表达网络已被用于获取除淀粉样蛋白 ß 和 tau 之外对 AD 重要的蛋白质和生物途径。然而,据我们所知,没有人尝试使用图神经网络和共表达网络来预测 AD 风险。
数据
为了证明如何使用蛋白质表达水平来预测诊断,我们使用了参与者基线访视期间收集的 ADNI 血浆数据。ADNI 是一项长达数十年的纵向多中心研究,完成后将招募近 2,000 名受试者。它包含来自受试者的丰富表型数据,包括有关临床状态、神经影像学、生物标志物、认知和遗传图谱的信息。本教程中的数据包含大约 190 种蛋白质的表达数据,这些数据收集自 58 名健康的老年人、395 名轻度认知障碍 (MCI) 患者和 112 名阿尔茨海默病患者(总共 565 名受试者)。在我们的数据集中,蛋白质分析物是列,受试者是行,每个细胞代表给定受试者的蛋白质表达水平。
经过一系列预处理步骤(包含在附录:附加信息中),我们创建了一个反映蛋白质之间双权重中相关(一种依赖于中位数而不是平均值的相关性,通常用于蛋白质组学分析)的矩阵。这将作为我们图结构的加权邻接矩阵,因此称为“蛋白质共表达矩阵”或加权邻接矩阵。
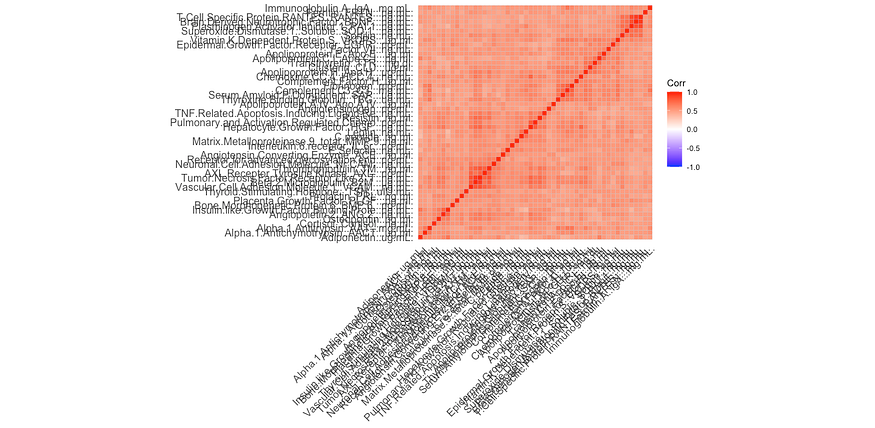
邻接矩阵的可视化
我们的 GNN 实现利用了这个加权邻接矩阵、蛋白质名称列表、每个受试者的诊断以及用于制作邻接矩阵的对数转换回归表达值。出于本教程的目的,我们将诊断二进制化为“1”(如果为 AD),否则为“0”。此代码可以轻松更新以适应多级分类(并纳入 MCI 诊断)。下面显示了编译的患者水平数据(由 S 显示)和诊断 (y) 的结构示例。

矩阵 S 中的患者水平蛋白表达数据示例,以及向量 y 中的二元诊断标记。
我们的方法
我们方法的关键方面是将每位患者表示为图,以蛋白质为节点,以蛋白质表达水平为节点特征,并对他们的诊断进行图级预测。 所有单个患者图的边将具有相同的通用固定图结构,由上述加权邻接矩阵或蛋白质共表达矩阵定义。 这些患者图中会有所不同的是节点值。每个患者图的节点值将由该给定患者的单个蛋白质表达水平表示,即由上面显示的主题矩阵 S 的一行的值表示。
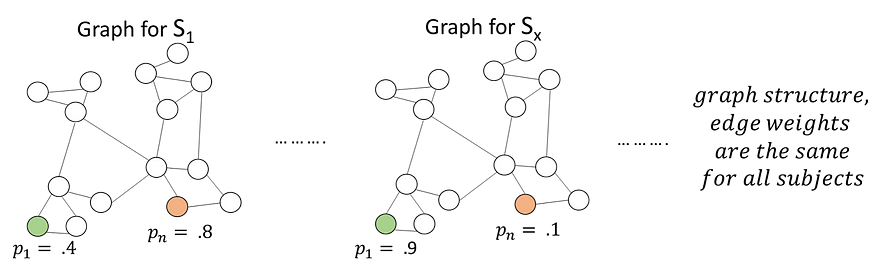
图级别或患者级别预测任务的数据结构可视化
这种对所有患者使用固定图的方法似乎不合常规。然而,一个指导性的假设是,在节点群落中会出现一些结构,其中一些相连的蛋白质“群落”在阿尔茨海默病中的作用可能比其他“群落”更重要。因此,这些蛋白质“群落”具有高表达值的患者可能更有可能产生阳性诊断。
为了做出考虑到社区结构的图级预测,我们将使用分层聚类,如下图所示。
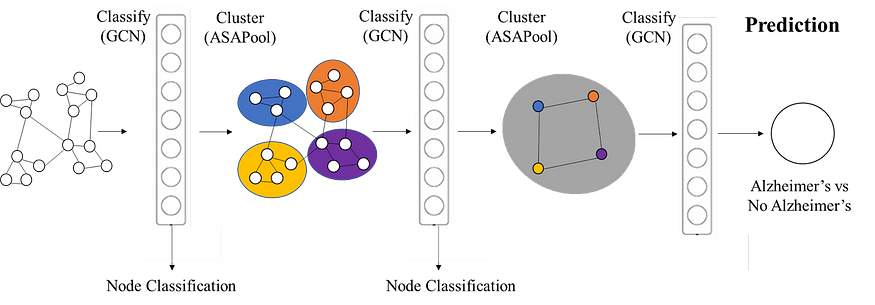
我们的模型架构概述
我们注意到我们的图不应该是排列不变的,因为不同特定节点的特征很重要。这与 GCN 的排列不变性质形成鲜明对比。为了包含位置编码,我们将随机生成的长度为 3 的浅编码特征向量添加到每个节点的节点嵌入中。这种浅层编码在所有主题图中也保持不变。因此,每个节点的特征是标量蛋白表达水平(因图而异)和位置编码(在图中保持不变)。
实现理论
我们正在使用图卷积网络 (GCN) 来学习每个节点的本地网络信息,并相应地生成节点嵌入。GCN 依赖于消息传递方法,其中节点交换发送“消息”的信息。然后,这些消息以某种方式被 “聚合” 或组合起来,以学习节点的新嵌入。这个过程是置换等变的,这意味着如果我们混淆节点顺序,结果将保持不变。为了保持不同蛋白质的一些编码,我们手动为每个蛋白质添加了一个随机向量的浅编码,该编码在所有图中保持不变。
如果我们想进行图级预测,我们想对所有节点信息进行一些聚合。但是,对于简单的平面聚合,例如所有节点值的平均值或所有节点值的最大值,有关图中细微之处的重要信息会丢失。为了解决这个问题并捕获本地结构,我们使用分层池,其中池化发生在不同的阶段。通过聚合来自具有相似 GCN 嵌入的节点的信息,然后应用非线性转换,可以捕获更精细的信息。在实践中,我们通过训练 GCN 嵌入,然后使用 ASAPool 方法进行聚类,并重复此过程来实现这一点(Ranjan 等人)。
实施演练
加载和预处理数据:
我们首先将邻接矩阵加载为 NumPy 对象,然后使用 NetworkX 的 nx.from_numpy_matrix 命令将其转换为图(G)。我们还创建了一个蛋白质名称的字符串列表,并使用 nx.set_node_attributes 将这些 设置为 node 属性。我们还从预处理的表达式值创建一个 565 x 51 的 NumPy 对象。我们将此、二值化诊断列表和邻接矩阵转换为 PyTorch 张量;我们还使用 torch_geometric.utils.from_networkx 将 NetworkX 图G 转换为 PyTorch Geometric 对象。
然后,我们创建一个字典 (split_idx),分别记录将用于训练集、验证集和测试集的索引。对于每个受试者,我们捕获包含所有 51 种蛋白质表达水平的行向量,并将其转置到列向量中,将其存储为 x。我们还捕获每个主题的诊断信息,并将其存储为 y。如果主题的索引位于我们的 split_idx 字典中对应于 “train” 的值列表中,我们会将描述同质图的数据对象附加到 train_list。此数据对象包含 x 作为我们的节点特征矩阵和 y 作为我们的真实图级标签。
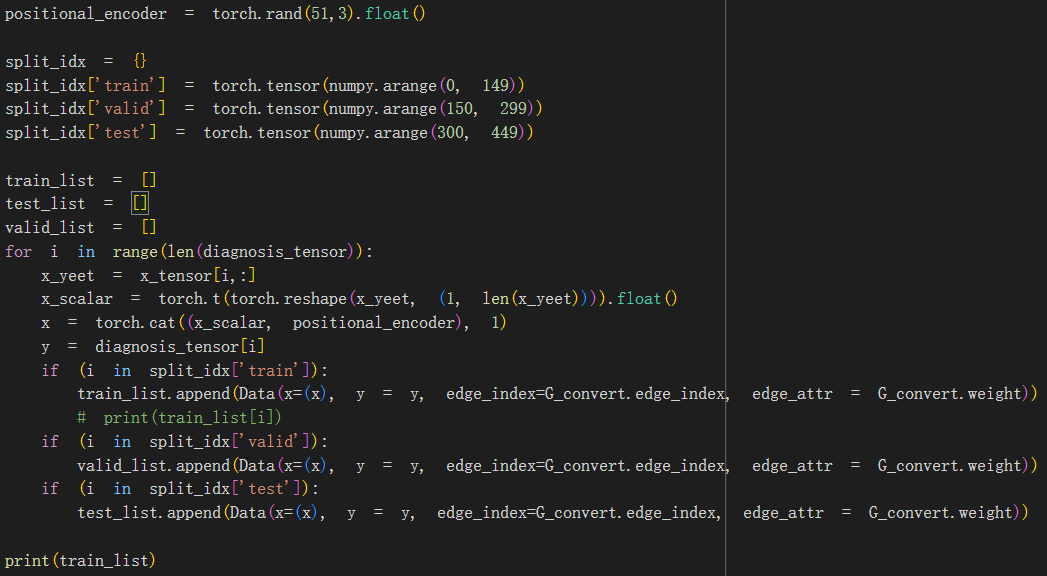
此外,它还从 G 捕获边缘索引和权重,并将其存储为 edge_index 和 edge_attr 中每个单独的数据对象。换句话说,对于每个主题,我们都会创建一个图(存储在 PyTorch Geometric 数据对象中),其中节点是蛋白质,特征是表达水平;边和权重在所有主体中都是固定的,因为它们取自邻接矩阵(转换为图对象 G)。每个图将分类为一个诊断类别(1 表示 AD,否则为 0)。受试者的图将根据受试者的索引附加到 train_list、valid_list 或 test_list。我们将这些单独的列表存储到不同的 PyTorch DataLoader 中,允许我们以大小为 32 的批次传递样本。
GCN 实施:
我们构建了第一个图卷积网络来进行节点级预测,共有五层,包括 256 维隐藏层。以我们的特征向量 x 和存储在图 G 中的边缘索引列表为例,我们将批量归一化、修正线性单元函数(我们的非线性)和以 50% 的概率退出应用于除最后一层之外的所有层。对于最后一层,我们应用一个 softmax 函数(将这个线性层的输出转换为分类概率分布),然后进行对数变换。
然后,我们使用模块 ASAPool 将节点分组到集群中。简而言之,ASAPool 最初根据其 1 跳邻域为所有节点创建集群,其中每个节点都是其自身集群的中介。集群中节点的成员资格是使用自注意力机制确定的,其中对集群中所有组成节点的查询提供注意力分数,该分数定义集群中给定节点的成员资格强度。然后使用基于图卷积方法的适应度分数选择性能最佳的集群,以捕获局部最大值/最小值。为池化图选择一小部分最适合的集群,并通过考虑池化集群是否包含任何公共节点,或者集群中的任何组成节点是否是原始图中的邻居来计算新的边权重。更多详细信息可以在原始论文 (Ranjan et al) 中找到。
在我们的模型中,我们使用 50% 的池化比率;换句话说,我们选择池化图中前 50% 的最佳拟合集群。对于另一个 GCN 分类和池化周期,重复此作,然后是最终 (GCN) 层。然后使用全局均值池化来执行图级聚合,并将线性变换应用于此图级预测,从而为我们提供最终的模型输出。
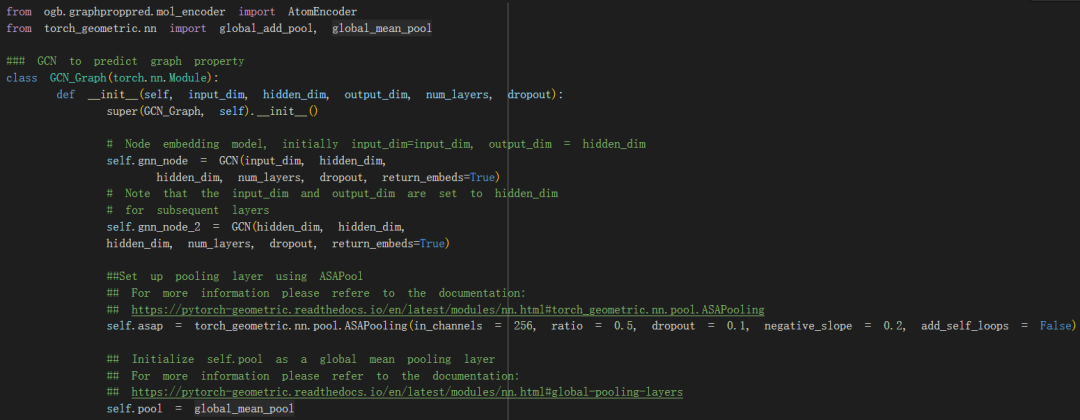
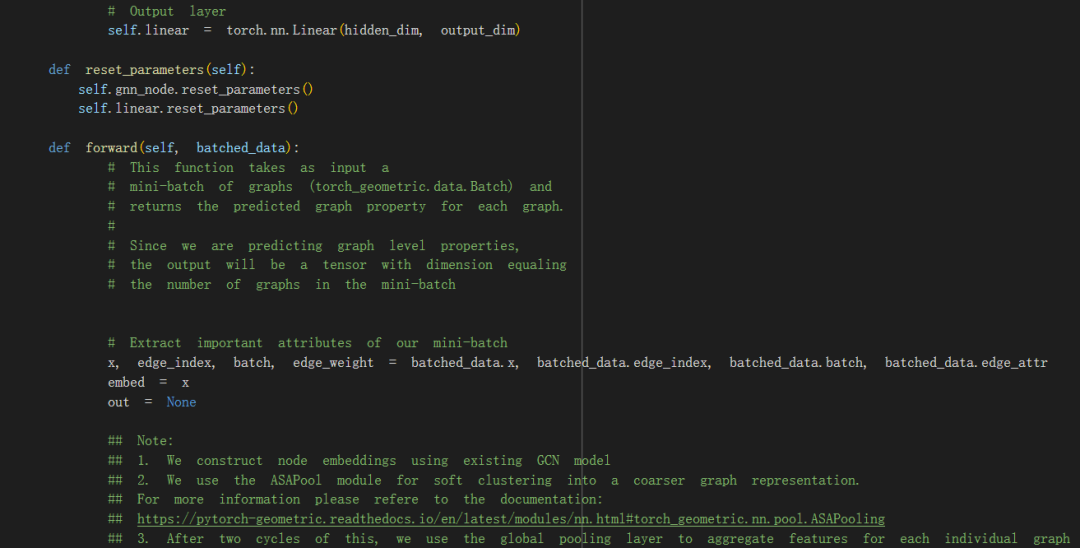
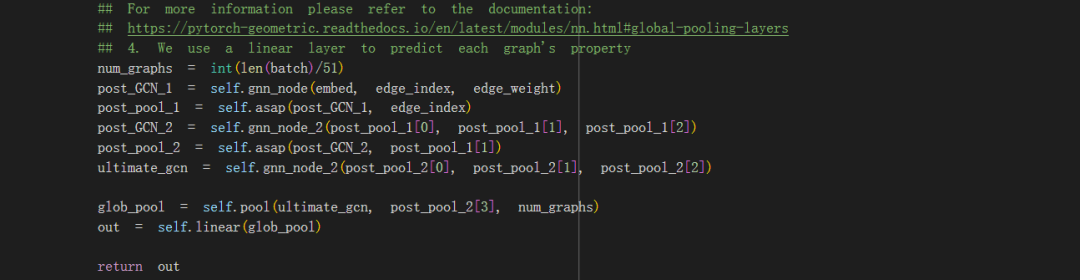
这些 GCN 和 ASAPool 层构成了我们的模型架构。在训练模型时,我们获取每个批次并将优化器的梯度设置为零。我们为每个批次运行模型,并使用 torch.nn.BCEWithLogitsLoss 计算损失。然后,我们分别使用 loss.backward() 和 optimizer.step() 反向传播错误并更新模型参数。当我们循环遍历每个批次时,我们会将 True 诊断标签和我们的模型输出记录在两个单独的列表中。我们将这些组合在一个字典中,y_true 和 y_pred 作为键。
性能
我们利用来自 “ogbg-molhiv” 数据集的 ROC-AUC 评估器来评估我们模型的性能。这构建了一个接收者作员特征曲线,该曲线绘制了真阳性率与假阳性率的关系;该曲线下的面积衡量模型在两个诊断组(在我们的例子中为 AD 或其他)之间的预测能力。在每个 epoch 中,我们记录训练、验证和测试组的训练损失和准确性(由 ROC-AUC 评估器比较我们的 y_true 和 y_pred 值计算的准确性)。我们以 0.001 的学习率运行了 50 个 epoch;最后,我们的训练集准确率为 50.15%,验证集准确率为 49.77%,测试集准确率为 49.77%。
我们将这种性能与基本的普通最小二乘模型进行了比较,其中我们的 X 是蛋白质表达值的预处理矩阵,y 是每个受试者的诊断列表。我们创建了一个 52 1 权重向量 (),表示每种蛋白质(即回归模型中的唯一参数)对诊断结果的权重。我们模型的输出是 X;我们将这个向量和 y 向量转换为 PyTorch 张量,并使用 torch.nn.BCEWithLogitsLoss 再次计算损失。我们使用一个简单的阈值来测量准确性,其中小于 0 的预测值将被分配为 0,否则将被分配为 1。这个 OLS 模型的损失为 0.7197,测试集的准确率为 28%。
讨论和进一步改进
我们注意到,我们的模型在测试集上的表现明显优于最小二乘模型 (28%)。未显示的是没有分层结构的单个 GCN 的性能,其性能相似,为 25-30%(包括我们将 GCN 更改为 GAT 等其他模型时)。这表明与朴素的平面模型相比,分层模型具有显著的优势。
然而,鉴于测试、训练和验证数据集的准确率为 ~50%,我们的模型还有很大的改进空间。这可以通过使用 GCN 以外的基础模型来生成节点嵌入来实现,包括图注意力网络,它可能在我们的蛋白质网络当前表示的完全连接的图结构上运行良好。也可以使用图同构网络 (GIN),它在结构区分方面更具表现力。
数据的预处理和存储方式也有改进的空间。例如,我们可以对邻接矩阵中的边进行阈值设置,并将边缘丢弃到低于某个阈值,以避免图如此连接。我们还可以更明确地解释边缘权重在我们的处理中的作用,目前仅由内置函数解释,在我们的实现中没有明确处理。
搜索超参数空间或更改模型配置(例如聚类程度、聚类周期数)也可能是有益的。
考虑我们对所有患者的固定图结构进行作的方法是否存在根本性限制是很有趣的。鉴于图中唯一发生变化的特征是每个节点的单个标量特征,因此可能没有足够的表达能力来区分患者并准确分类疾病。对这一点的系统调查可能会引起人们的兴趣。也许可以通过根据 ADNI 中的其他数据(例如神经成像数据)为每位患者添加节点嵌入来增强数据集,以提高数据的丰富性。
附录:更多细节
数据准备
我们首先通过去除一定比例的样品低于最小可检测剂量 (LDD) 的蛋白质来预处理数据。最低可检测剂量是每种蛋白质的最低可靠测量值(每种蛋白质的唯一值)。如果给定蛋白质的大部分样品低于 LDD,那么我们就会对该蛋白质测量的可靠性完全失去信心。我们按照 ADNI 生物标志物联盟制定的方法使用了 25% 的阈值;如果给定蛋白质的样品中有超过 25% 低于 LDD,则将其从进一步分析中剔除。否则,我们将低于 LDD 的值替换为实际的 LDD 级别。这给我们留下了 52 种蛋白质。然后,我们对蛋白质表达水平进行对数转换,使蛋白质间的变化处于相似的数量级内(见图 1)。我们最后的预处理步骤是回归可能影响蛋白质表达水平的协变量的影响。我们希望我们的蛋白质共表达网络能够有效地捕捉蛋白质之间的关系,而这些估计可能会被年龄、性别和诊断的影响所掩盖。
引用
Thomas N. Kipf, Max Welling, Semi-Supervised Classification with Graph Convolutional Networks, https://arxiv.org/abs/1609.02907
Rex Ying, Jiaxuan You, Christopher Morris, Xiang Ren, William L. Hamilton, Jure Leskovec, Hierarchical Graph Representation Learning with Differentiable Pooling, https://arxiv.org/abs/1806.08804
Ekagra Ranjan, Soumya Sanyal, Partha Pratim Talukdar, ASAP: Adaptive Structure Aware Pooling for Learning Hierarchical Graph Representations, https://arxiv.org/abs/1911.07979



内容中包含的图片若涉及版权问题,请及时与我们联系删除
