
近日,南开大学统计与数据科学学院郑伟教授团队,与新加坡国立大学张阳教授在Nature Biotechnology上发表了题为《Deep-learning-based single-domain and multidomain protein structure prediction with D-I-TASSER》的论文。这项研究工作提出了一种融合深度学习与基于统计物理能量函数力场的片段组装结构预测方法D-I-TASSER,用于原子级蛋白质结构预测。D-I-TASSER还引入了结构域划分与组装模块,实现了大型多结构域蛋白质的自动预测。基准测试显示,D-I-TASSER在单结构域与多结构域蛋白质结构预测问题中,结果均要优于DeepMind公司的诺奖算法AlphaFold2和AlphaFold3。此外,在第15届世界蛋白质结构预测大赛(CASP15)中,D-I-TASSER算法在单结构域和多结构域蛋白质结构预测任务中均获得冠军。南开大学郑伟教授为该论文的第一作者,密歇根州立大学乌云其其格博士以及新加坡国立大学李阳特别研究员为共同第一作者。新加坡国立大学张阳教授和密歇根大学Lydia Freddolino副教授为论文的共同通讯作者。

背景介绍
蛋白质三维结构预测(也称“蛋白质三级结构预测”)是指基于氨基酸序列推测其三维空间结构的过程。由于蛋白质只有在折叠成特定的空间构象后才能具备相应的活性和生物学功能,因此结构预测对于理解蛋白质功能、揭示疾病机制以及进行药物设计具有重要意义。传统的结构解析依赖于X射线晶体学、核磁共振等实验技术,不仅耗时长、成本高,还对实验条件要求苛刻。近年来,该领域的一个重要里程碑是使用深度学习预测空间几何约束结构特征,例如接触图(contact map)、距离图(distance map)、氢键网络(hydrogen bond network)和扭转/二面角(torsion/dihedral angle),然后通过构建几何约束的能量函数,并采用能量最小化策略,进而建立蛋白质全长结构的预测模型。随后,AlphaFold2引入了端到端学习框架,直接根据序列和共进化信息预测蛋白质的结构,极大地提高了预测精度和效率。最新的AlphaFold3进一步结合扩散模型,使得端到端学习的效果和泛化能力进一步增强。这一系列深度学习方法的出现,使得蛋白质结构预测准确率远远超过传统结构折叠方法(例如I-TASSER、Rosetta和QUARK)。基于物理的传统折叠方法在揭示蛋白质折叠原理与动力学机制方面具有不可替代的重要价值。然而,在实际结构预测任务中,物理模拟方法是否仍具必要性,以及深度学习方法能否完全取代传统物理建模手段,仍有待进一步探讨。
目前蛋白质结构预测问题仍面临一个重要挑战,即大多数先进方法主要聚焦于单个结构域(single domain)的预测。结构域(domain)是复杂蛋白质三级结构中的基本折叠和功能单元,大约60%的原核生物蛋白质和80%的真核生物蛋白质都由多个结构域(multi-domain)组成,并且依赖结构域间的相互作用执行更复杂的生物学功能,因此在结构上更为复杂(图1)。目前,无论是基于物理的结构预测方法,还是基于深度学习的结构预测方法,在处理多结构域蛋白质结构预测问题时都缺乏有针对性的多结构域处理模块。因此,如何准确高效地预测多结构域蛋白质结构,仍然是亟需解决的关键科学问题。
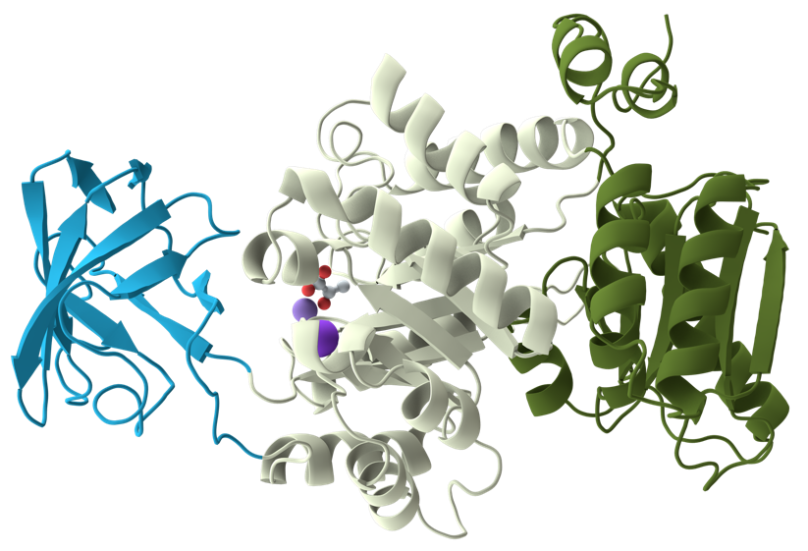
图1. 多结构域蛋白质单体示意图。该蛋白质包含3个结构域,不同结构域被标记为不同颜色。
在这项研究中,郑伟团队在张阳教授开发的I-TASSER统计物理能量方法的基础上提出了一种整合深度学习的预测方法D-I-TASSER。D-I-TASSER结合了深度学习与统计物理能量函数,采用副本交换蒙特卡洛模拟(REMC)进行蛋白质结构预测。同时,还引入了一个全新的结构域划分与组装模块,用于自动预测大型多结构域蛋白质结构。基准测试和CASP15盲测结果均表明,D-I-TASSER在结构预测的准确性上明显优于传统的I-TASSER系列方法,以及Google子公司DeepMind团队开发的诺奖算法AlphaFold2、AlphaFold3。
方法简介
D-I-TASSER方法能够有效处理单结构域蛋白质和多结构域蛋白质结构预测问题。对于单结构域蛋白质(图2),D-I-TASSER首先使用DeepMSA2深度序列比对算法,迭代搜索基因组(UniProt、UniRef等)和宏基因组(Metaclust、IMG/M,BFD等)蛋白质序列数据库,构建深度多序列比对(MSA)。随后,利用LOMETS3模板检索算法,快速识别与目标蛋白结构相似的模板,并生成相应的模板比对。接着,使用DeepPotential、AttentionPotential、AlphaFold2等深度学习模块预测接触图(contact map)、距离图(distance map)、氢键网络(hydrogen bond network)等空间约束。最后,在深度学习空间约束与统计物理能量函数共同构成的力场驱动下,采用REMC蒙特卡洛模拟算法进行蛋白质结构预测。

图2. D-I-TASSER单结构域蛋白质结构预测流程。
对于多结构域蛋白质(图3),D-I-TASSER能够成功地预测其结构,得益于其引入了一个全新的结构域划分与组装模块,该模块包含5个关键步骤:
(1)预测结构域边界;
(2)生成结构域级特征;
(3)构建全链MSA与空间约束;
(4)收集全链模板;
(5)全链级结构预测。
具体地,首先使用ThreaDom和FUpred预测目标蛋白质的结构域边界,并将目标蛋白质划分为多个结构域。随后,为每个结构域生成结构域级的MSA、模板和空间约束。在此基础上,将原始的全链MSA与结构域级MSA结合,通过棋盘式对齐方式构建全链级MSA。将全链的 MSA 输入至深度学习模块,以获取全链级别的空间约束,并进一步与各结构域预测所得的空间约束信息融合,最终构建出完整的全链空间约束体系。全链级模板则包含DEMO2将各结构域模板拼接成的覆盖全链的模板,以及全链级LOMETS3模板。最后,通过全链的D-I-TASSER组装模拟,在混合的结构域级和跨域空间约束的引导下,构建多域结构模型。
D-I-TASSER与以往的多结构域结构预测算法I-TASSER-MTD的不同之处在于,之前的算法尝试将多结构域蛋白质分割后独立预测每个结构域的结构,然后再进行拼装。这样做的缺点是结构域内约束和结构域间约束可能会冲突,导致预测结果不稳定。D-I-TASSER采用的是结构域级的特征拼装,再将拼装好的完整的全链级的特征输入到深度学习模型、模板检索算法或者结构预测模块,进行整体预测,这样能够最大程度地减少结构域内约束和结构域间约束的冲突,提升预测精度。
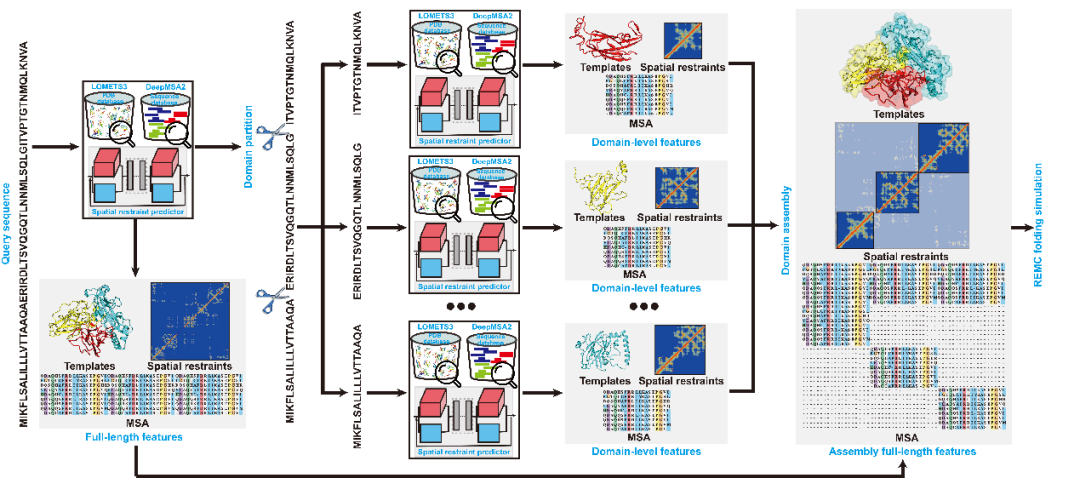
图3. D-I-TASSER多结构域蛋白质结构预测流程。
结果分析
(一)单结构域蛋白质
预测性能
这篇文章首先评估了D-I-TASSER在单结构域蛋白质上的预测性能(图4)。在500个非冗余“困难”结构域测试集上(困难蛋白质指的是没有同源模板的蛋白质),D-I-TASSER展现出了对AlphaFold2的全面优势。具体来看,D-I-TASSER的平均TM-score为0.870,显著高于AlphaFold2的预测结果(TM-score=0.829,相对提升5.0%),且在84%的目标上生成了质量更高的预测结果。对于特别困难的148个结构域(图4红色标注区域),D-I-TASSER表现大幅优于AlphaFold2(TM-score=0.707 vs 0.598)。其中,D-I-TASSER 在 63 个蛋白质目标上的预测精度优于 AlphaFold2,且差异超过 0.1 分;而 AlphaFold2 仅在 1 个目标上展现出相对优势。
注:TM-score是由张阳实验室首先提出、现已成为蛋白质结构相似性评估的主流指标。该分数范围在0到1之间,值越高表示预测结构与真实结构越接近。

图4. D-I-TASSER与AlphaFold2在单结构域蛋白质上的性能比较。
为了更全面地评估模型性能,文章比较了D-I-TASSER与不同版本的 AlphaFold 算法的预测精度,结果显示D-I-TASSER的预测性能优于所有版本的AlphaFold。D-I-TASSER能够成功地进行蛋白质结构预测,得益于其整合了多种特征,包括深度多序列比对、多来源的深度学习空间约束预测器、融合深度学习及统计物理能量函数力场的分子动态模拟等。图5对这些特征的贡献进行了系统性分析,随着不同类型约束的逐步融合,预测模型的TM-score持续提升。最终,整合全部深度学习空间约束的D-I-TASSER模型的TM-score达到0.870,超越AlphaFold2(0.829)和AlphaFold3(0.849),这凸显了多源深度学习约束整合带来的优势。
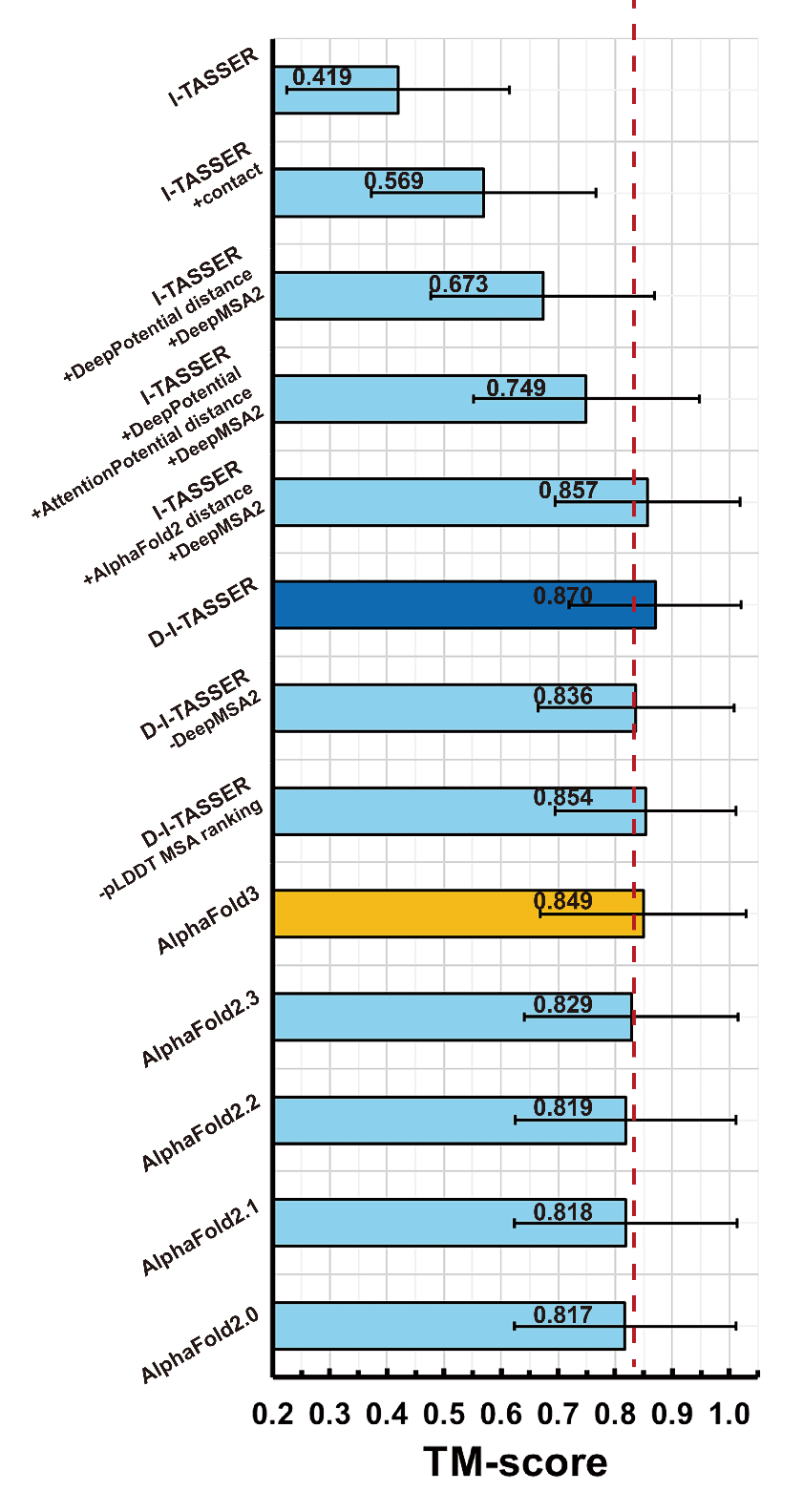
图5. D-I-TASSER与AlphaFold全系列、不同约束条件下的I-TASSER体系的比较。
值得强调的是,高质量多序列比对的构建对D-I-TASSER蛋白质结构预测的精度,同样起到了至关重要的作用。消融实验表明,移除DeepMSA2模块后,模型平均TM-score从0.870降至0.836(p-value=3.63E-69)。DeepMSA2通过丰富的宏基因组数据库和深度学习驱动的MSA筛选算法,显著提升了预测效果。即使将DeepMSA2的MSA直接用于AlphaFold2,也能小幅提高预测准确率(0.819→0.841),但整体性能仍不及D-I-TASSER(0.870)。这进一步验证了D-I-TASSER的卓越性能得益于多种深度学习空间约束及统计物理能量函数的有效融合。
为了验证DeepMSA2与多源深度学习空间约束在结构预测中的作用及其协同效应,文章以来自番茄丛枝矮化病毒的RNA沉默抑制因子p19(PDB ID: 4jgnA)为例(图6),深入剖析了不同模块对建模质量的影响。由于该蛋白质缺乏足够的同源序列,AlphaFold2构建的MSA较浅(Neff=0.36),因此其预测的结构质量较差(TM-score=0.335;图6A),其预测的距离图也存在较高误差(MAEn=3.20Å;图6C)。引入DeepMSA2后,MSA的比对深度提升了6.75倍,显著改善了AlphaFold2预测的距离图精度(MAEn=0.69Å;图6D)。然而,AlphaFold2的距离图仍然缺乏N端与其他区域之间的距离信息。D-I-TASSER进一步结合DeepPotential和AttentionPotential模型后,预测的距离图覆盖了整个序列区域,整体准确性进一步提高(MAEn=0.45Å;图6E)。依托这一复合距离图,D-I-TASSER最终预测了一个高质量的结构(TM-score=0.871;图6B)。该案例表明,DeepMSA2构建的高质量MSA有效增强了进化特征的提取,而多源深度学习空间约束则进一步提升了空间信息预测的准确性与预测质量,两者的协同作用,是D-I-TASSER优异预测性能的关键所在。
注:MAEn是深度学习模型预测距离与实验结构的相应距离之间的平均绝对距离误差。

图6. 番茄丛枝矮化病毒RNA沉默抑制因子p19上AlphaFold2与D-I-TASSER的预测结果比较。
为了揭示D-I-TASSER如何在深度学习空间约束与统计物理能量函数的共同作用下实现高精度蛋白质结构预测,文章分析了其在磷酸合酶(PDB ID: 3fpiA)预测过程中的构象折叠轨迹(图7)。在深度学习空间约束与统计物理能量函数的引导下,在REMC模拟的前40轮分子模拟中,预测结构的质量迅速提升:MAEm从7.7 Å快速下降至1.2 Å,同时TM-score从0.31跃升至0.71。随着模拟的进一步推进,预测构象在第100轮时达到收敛状态,MAEm和TM-score分别稳定在0.39 Å和0.96的高精度水平。这一轨迹分析表明深度学习与统计物理能量函数的结合能够有效揭示蛋白质的折叠过程。
注:MAEm是深度学习模型预测距离与D-I-TASSER模型相应距离之间的平均绝对距离误差。

图7. D-I-TASSER在磷酸合酶(PDB ID: 3fpiA)结构预测过程中的构象折叠轨迹。
(二)多结构域蛋白质
结构预测能力评估
为了系统评估D-I-TASSER在多结构域蛋白质结构预测方面的能力,文章构建了一个包含230个多结构域蛋白质(共557个结构域)的基准测试集。图8A和8B分别总结了D-I-TASSER与AlphaFold2在全链级别和结构域级别的结构预测性能比较。结果表明,D-I-TASSER预测的全链级别和结构域级别模型的TM-score分别为0.720和0.858,较AlphaFold2相应模型的TM-score(0.638和0.835)分别提高了12.9%和2.8%。进一步分析表明,该性能优势主要体现在难度较高的预测目标(图8A和8B所示的红色标注区域)上。在这些困难目标上,D-I-TASSER相对于AlphaFold2的TM-score提升分别为17.1%(全链级别)和9.9%(结构域级别),凸显了其在复杂结构预测中的显著优势。

图8. D-I-TASSER与AlphaFold2在多结构域蛋白质上的性能比较(全链和结构域)。
D-I-TASSER主要通过两个方面来提升多结构域蛋白质结构预测的质量:(1)通过结构域划分策略,提升结构域级别预测结构的质量;(2)通过结构域特征组装策略,提升结构域间空间约束质量及全链级别预测结构的质量。
在 D-I-TASSER 提升结构域级别预测结构质量方面,文章分析了 C. reinhardtii 鞭毛辐射辐条蛋白质(PDB ID: 7jtkB)这一双结构域蛋白质的预测案例(图9)。由于检测到的有效序列数量较少(Neff=0.1),AlphaFold2预测的全链模型质量较差(全链TM-score=0.425),且结构域内的预测效果也不佳。相比之下,D-I-TASSER通过结构域划分与组装模块,提高了MSA的覆盖度(Neff=0.4),同时分别为两个结构域检测到额外的688条和15条同源序列,显著改善了距离图预测准确性。在更准确的约束引导下,D-I-TASSER最终预测了高质量的结构模型(全链TM-score=0.934),两个结构域的TM-score分别为0.971和0.910,远优于AlphaFold2的结果。该案例表明,在有效序列稀缺的情况下,结构域划分与组装的 MSA 构建策略,能够显著提升结构域内的MSA质量及预测结构质量,进而提升全链模型预测准确性,这是 D-I-TASSER 能够克服多结构域挑战的关键因素之一。

图9. C. reinhardtii鞭毛辐射辐条蛋白质上AlphaFold2与D-I-TASSER的预测结果比较。
在D-I-TASSER提升结构域间空间约束质量方面,文章考察了 InaD-like 蛋白质(Inactivation No Afterpotential D,PDB ID: 6irdC)这一双结构域蛋白质。虽然 AlphaFold2 能较为准确地预测各结构域的局部构象(TM-score 分别为 0.894 和 0.930),但由于多序列比对(MSA)信息有限,其预测结构域间的空间排布严重错误,导致整体模型偏离真实构象,全链 TM-score 仅为 0.510(图 10上图)。相比之下,D-I-TASSER在划分结构域后,借助比对深度更深的MSA,通过全链和结构域的MSA组装,有效提升了预测精度(全链TM-score达0.890,图10下图)。该结果表明,引入的结构域特征组装模块可有效挖掘更丰富的进化信息,从而提升多结构域蛋白质结构域间的预测质量,进而提升结构预测的准确性,这是D-I-TASSER能够成功预测多结构域蛋白质结构的重要原因。
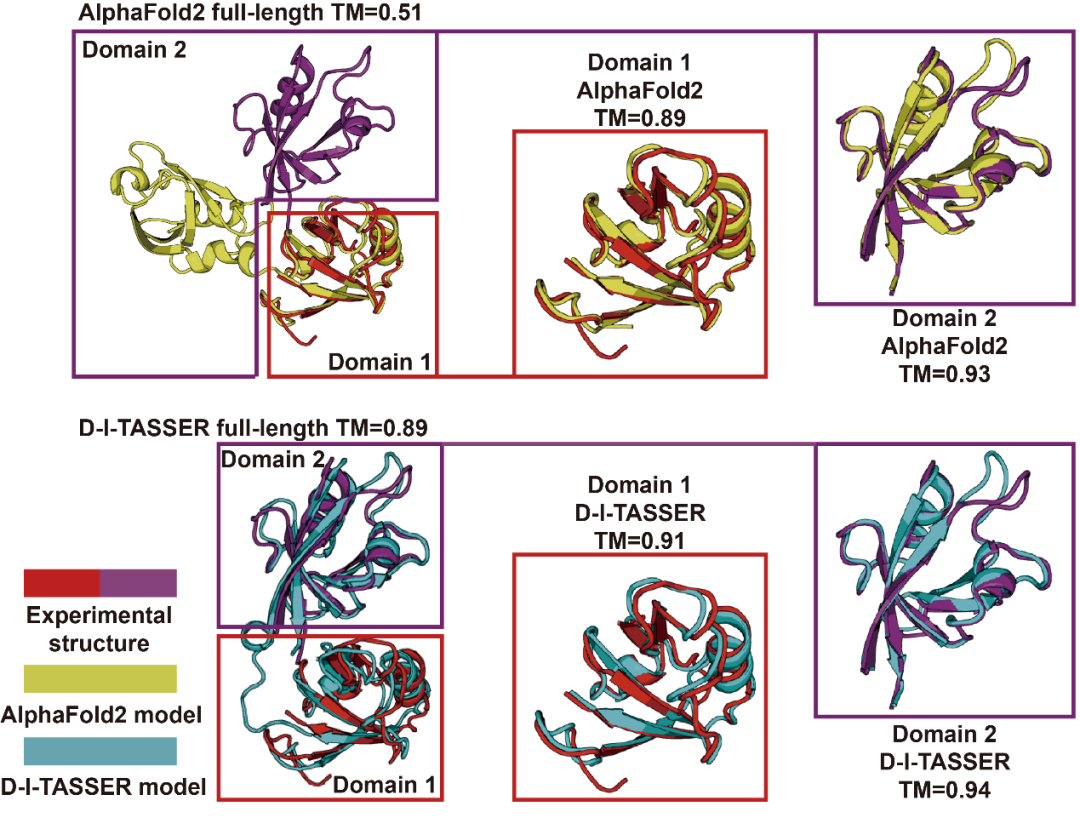
图10. 人类InaD-like蛋白质上AlphaFold2与D-I-TASSER的预测结果比较。
值得注意的是,多结构域蛋白质为了满足自身的功能需求,通常会在结构域方向上呈现多种构象变化。D-I-TASSER结合了深度学习空间约束与统计物理能量函数,通过REMC方法模拟生成了大量多样化的构象(decoys),为预测多构象状态的蛋白质提供了巨大潜力。图11展示了一个典型案例——新冠病毒(SARS-CoV-2)刺突蛋白质三聚体,其蛋白质链可以处于开放(open)或闭合(closed)两种构象状态(图11A)。这两种状态的主要差异来源于,与C端受体结合的结构域(RBD)相对于其他结构域发生了方向变化。D-I-TASSER成功预测了这两种构象(图11B),其中,第一个模型对应闭合状态(TM-score=0.940),第二个模型对应开放状态(TM-score=0.990)。如图11C所示,D-I-TASSER生成的构象大致可以分为开放、闭合和中间状态三类。SPICKER聚类分析显示,这些构象可以被聚类为五个簇,第一个模型(闭合状态)和第二个模型(开放状态)分别来自其中最大和次大的簇。相比于通常只生成单一静态模型的纯深度学习方法,图11所示结果突出体现了整合深度学习及统计物理能量函数的D-I-TASSER,在捕捉蛋白质多构象状态方面的内在优势。

图11. D-I-TASSER成功预测出新冠病毒(SARS-CoV-2)刺突蛋白质的开放和闭合两种构象状态。
(三)CASP15大赛
盲测综合表现
D-I-TASSER算法(参赛名:UB-TBM)参加了第15届世界蛋白质结构预测大赛(CASP15),并在单结构域预测(Regular)和多结构域预测(Inter-domain)比赛中均排名第一(图12A和12B)。CASP是国际公认的世界级蛋白质结构预测权威竞赛,每两年举行一次,旨在通过对大分子和复合物结构预测计算方法进行严格的评估,来推动结构预测领域技术的进步。CASP采用严格的双盲预测机制,长期以来被视为结构预测领域的“金标准”,并被业界誉为“蛋白质结构预测的奥林匹克竞赛”。郑伟教授团队在过去四届CASP比赛中均取得优异成绩,累计获得十项冠军。

图12. CASP15世界蛋白质结构预测大赛单结构域(A)和多结构域(B)预测比赛排名。
按照CASP15组委会公布的官方Z-score评分结果,D-I-TASSER的整体表现优于其他所有参赛组。在单结构域和多结构域目标中,D-I-TASSER的累计Z-score分别为67.20和35.53,显著高于AlphaFold2的32.05和2.11。值得注意的是,CASP15包含两个赛道:服务器赛道要求预测模型在72小时内提交;而人工赛道允许专家手动干预,并为每个目标预留最长三周的预测时间。即便在与人类专家组同场竞争的情况下,D-I-TASSER服务器在单结构域预测目标中依然取得了第二名(若按Z-score>0.0统计则为第一名)的优异成绩。此外,在多结构域预测中,D-I-TASSER服务器的表现明显优于包括人类专家组在内的其他所有参赛组,其累计Z-score比排名第二的参赛组(24.96)高出42.3%。
作为蛋白质结构预测领域的里程碑式方法,AlphaFold系列方法凭借其革命性的端到端深度学习框架,持续引领着单链结构预测的技术革新。然而在复杂多结构域及困难折叠单元的预测任务中,D-I-TASSER方法展现出更强的预测能力——如图13所示,通过对困难(FM)蛋白质结构域(50个)和多结构域蛋白质(20个)的TM-score对比分析,D-I-TASSER预测结果均显著优于包括AlphaFold3在内的所有AlphaFold系列方法。

图13. CASP15中困难(FM)蛋白质结构域和多结构域蛋白质上D-I-TASSER与AlphaFold全系列的TM-score比较。
文章选取了19个单结构域及8个多结构域目标作为代表性案例,其中D-I-TASSER相较于AlphaFold2的TM-score提升均超过0.15分差(图14)。值得关注的是,D-I-TASSER成功预测了多个大尺寸(残基数大于3,000)多结构域蛋白质的三维结构,例如T1169(残基数3,364)预测模型的TM-score达到0.800。这一系列成果标志着深度学习约束在大尺寸蛋白质结构预测领域取得了重要进展,突破了传统方法长期以来面临的挑战。
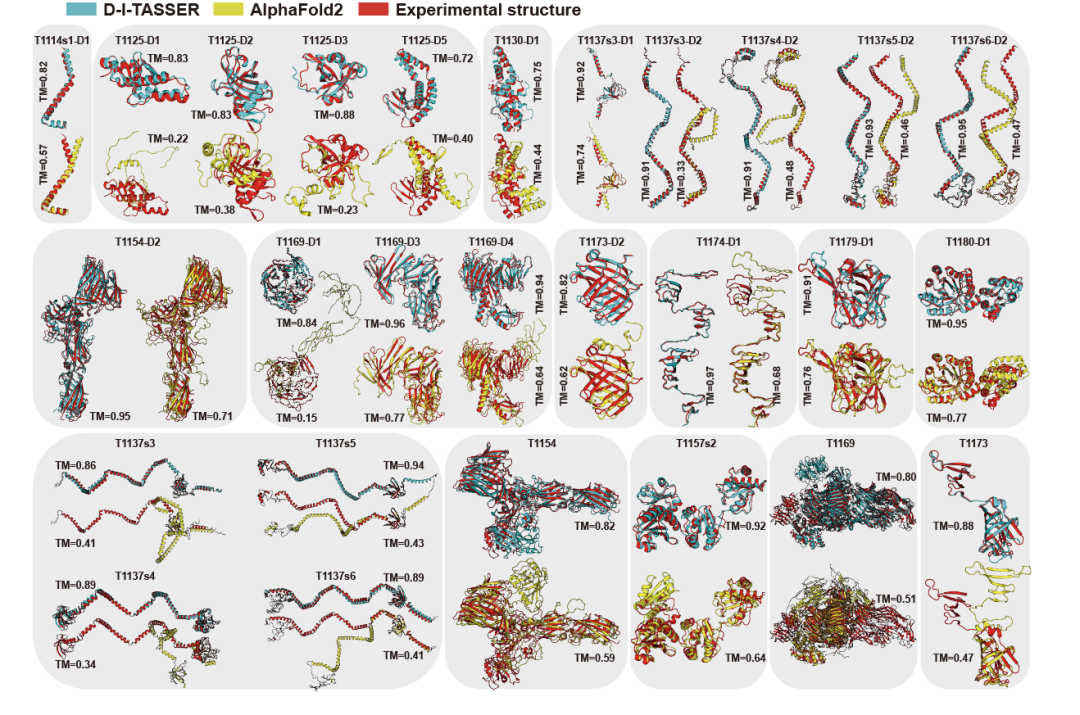
图14. CASP15中D-I-TASSER与AlphaFold2在部分靶标上的预测结果比较。
(四)大规模人类蛋白质组
结构和功能预测
D-I-TASSER对人类基因组的19,512个蛋白质和34,968个结构域进行了结构预测,涵盖了约95%的人类蛋白质组。为评估模型质量,D-I-TASSER设计了eTM-score作为预测精度的估计指标。结果显示,D-I-TASSER成功预测了80.5%的单结构域和72.8%的全链蛋白质结构(图15A),且预测结果与AlphaFold2数据库高度互补。值得注意的是,D-I-TASSER还准确预测了3,020个AlphaFold2难以可靠预测的人类蛋白质,凸显其在填补结构预测盲区方面的重要价值。进一步分析表明,预测结构的eTM-score在各染色体间分布均匀(图15B),证实预测精度与基因在染色体上的定位无显著相关性。此外,D-I-TASSER还对人类蛋白质组的生物学功能进行了注释(图15C),包括基因本体功能标签(GO)、酶分类(EC)以及小分子绑定位点(LBS)。
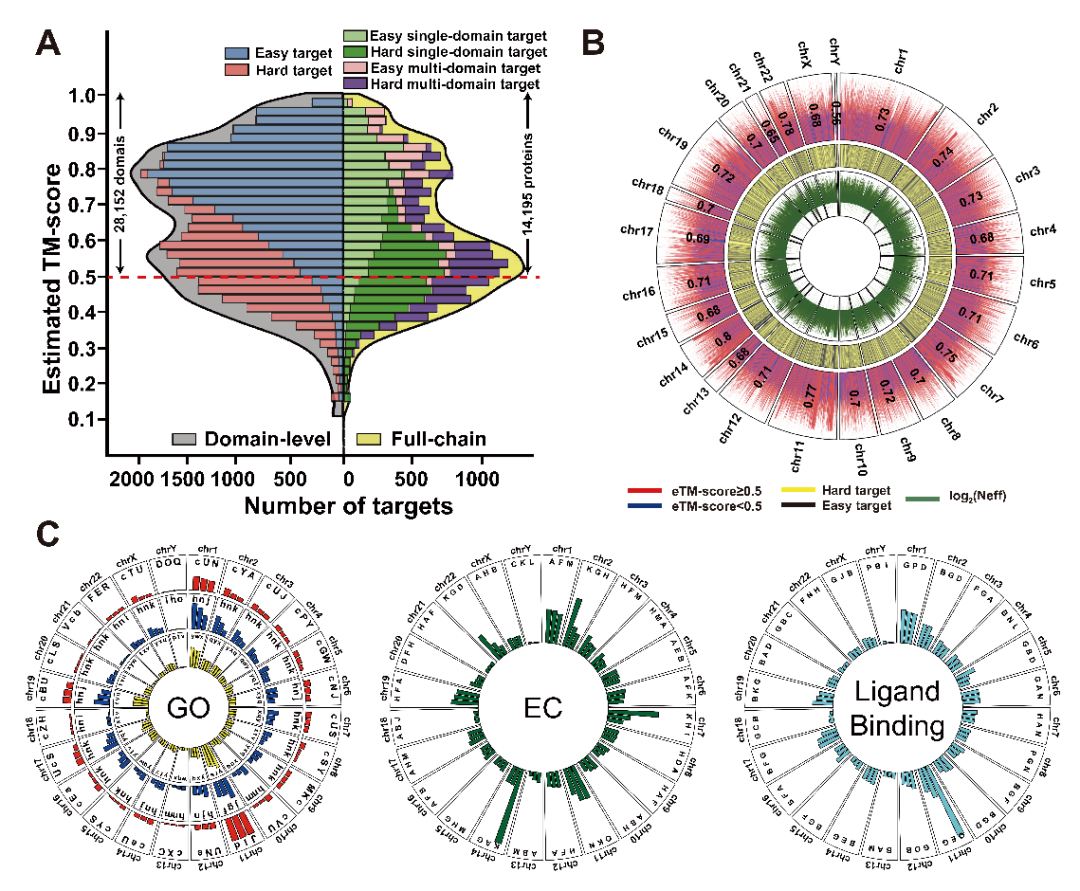
图15. (A) D-I-TASSER在人类蛋白质组上的eTM-score分布;(B) 对eTM-score分布(外道)、目标类型(中道)和Neff对数(内道)的染色体级别分析;(C) 基于 D-I-TASSER 的人类蛋白质组功能注释。
结论与展望
本研究开发了一个蛋白质结构预测算法D-I-TASSER,该方法通过整合多种深度学习空间约束及统计物理能量函数,并引入全新的结构域划分与组装模块,实现了大尺寸单结构域及多结构域蛋白质的自动化预测。大规模基准测试结果表明,D-I-TASSER在单结构域与多结构域蛋白质预测方面均显著优于主流的AlphaFold系列方法。D-I-TASSER的卓越性能得益于多种深度学习空间约束及统计物理能量函数的有效融合。新引入的结构域划分与组装模块可有效挖掘更丰富的进化信息,从而提升多结构域蛋白质预测的准确性。进一步地,在国际权威的CASP15盲测中,D-I-TASSER在单结构域和多结构域预测两个任务类别中均取得了最佳表现,验证了其在实际应用中的广泛适用性和领先水平。
总体而言,D-I-TASSER通过结合先进的深度学习技术、统计物理能量函数及自动化的多结构域处理模块进行结构预测,实现了高难度目标蛋白质和多结构域蛋白质预测精度的显著提升。并且D-I-TASSER算法框架具有良好的可扩展性,能够不断整合更高精度的深度学习方法,进一步应对孤儿蛋白质或蛋白质复合物结构预测问题中的挑战。
Nature Biotechnology是国际生物技术领域及计算结构生物信息学公认的顶级期刊,五年影响因子57.0,每年发文仅百余篇,而纯计算方法只占该杂志发文量的10%左右。本研究得到了南开大学前沿交叉学科研究院、传染病溯源预警与智能决策全国重点实验室、自然科学基金委等多家单位的支持。
作者及团队介绍
郑伟,南开大学统计与数据科学学院教授,博士生导师。其长期从事于基于深度学习及统计物理能量函数的生物分子及其互作的结构预测研究,主持开发了C-I-TASSER、C-QUARK、D-I-TASSER、DMFold等一系列结构预测算法,累计获得CASP国际大赛十项冠军,并多次受邀在国际会议作特邀报告。郑伟教授累计在Nature Biotechnology、Nature Methods、Nature Communications、PNAS等高水平SCI期刊发表文章50余篇,总引用3400余次。其主导开发的结构预测算法已经累计服务超过100个国家的近10万名用户。此外,南开统计在生物医学相关方向的研究涉及生物统计、生物信息学、数学流行病学、计算机视觉和自然语言处理在生物医学中的应用等,近年来在生物医学权威期刊Nature Biotechnology、Nature Cancer、Circulation、Gut、Nature Communications等权威杂志发表数十篇论文,多篇疫情研判报告获得国家领导人批示,在国际蛋白质结构和功能预测等竞赛中多年多次获得冠军。
乌云其其格,密歇根州立大学计算机科学与工程系博士,曾于南开大学获得生物信息学硕士学位和信息与计算科学学士学位。研究方向涵盖计算生物学、生物信息学、系统进化、蛋白质结构预测及其在疾病机制与药物研发中的应用。研究成果在Nature Biotechnology、Nature Methods、Nucleic Acids Research等国际权威期刊及会议发表多篇论文,总引用次数超过500次。
李阳,新加坡国立大学癌症科学研究所特别研究员及资深研究科学家,专注于深度学习在结构生物信息学中的研究与应用。
新加坡国立大学张阳教授为本文通讯作者,常年从事基于人工智能的蛋白质和RNA结构预测及序列设计研究,其团队开发的计算机算法连续9次获得CASP大赛冠军。密歇根大学Lydia Freddolino副教授为共同通讯作者。
参考资料
Zheng, W., Wuyun, Q., Li, Y. et al. Deep-learning-based single-domain and multidomain protein structure prediction with D-I-TASSER. Nat Biotechnol (2025).
https://doi.org/10.1038/s41587-025-02654-4

关注南开大学统计与数据科学学院生物信息学课题组,获取最新研究进展与成果分享。
内容中包含的图片若涉及版权问题,请及时与我们联系删除
