
DRUGAI
今天为大家介绍的是来自德国于利希研究中心(Forschungszentrum Jülich GmbH)和黑尔姆霍兹人工智能中央单位(Helmholtz AI Central Unit)的一篇论文。从一般序列、结构折叠或进化信息推断酶功能的现有工具通常都很成功。然而,如果局部结构特征的偏差影响了功能,这些工具可能会导致错误分类。在此,作者提出了TopEC,一种基于局部化3D描述符的3D图神经网络,用于从酶结构中学习酶的化学反应并预测酶委员会(EC)分类。通过使用消息传递框架,作者纳入了距离和角度信息,与常规2D图神经网络相比,显著提高了EC分类的预测性能(F score:0.72)。作者训练了没有结构折叠偏差的网络,可以对广泛的功能空间(>800个EC)中的酶结构进行分类。作者的模型对结合位点位置的不确定性以及不同结合位点中的相似功能具有稳健性。作者观察到TopEC网络从生物化学特征和局部形状依赖特征之间的相互作用中进行学习。TopEC可在GitHub上获取:https://github.com/IBG4-CBCLab/TopEC和https://doi.org/10.25838/d5p-66

蛋白质是所有细胞生命的基础。蛋白质数据库(PDB)是全球生物分子3D结构实验信息的存储库,到2021年底包含185,539个晶体结构,其中25,190个是本研究关注的非冗余酶结构。最近在蛋白质结构预测方面的发展极大地改进了酶的结构模型预测,并导致了大型结构数据库的出现。然而,对于国际生物化学与分子生物学联盟命名委员会(IUMBMB)提出的所有酶功能中,只有60%的酶功能有预测结构模型可用。因此,从(预测的)酶结构中准确注释酶的分子功能仍然具有挑战性。为许多序列实验性地确定酶功能非常耗时,通常酶功能不能直接从结构表示中推导出来,或者在数据库中错误地将酶功能注释到序列上。以酶结构作为输入的计算方法可以弥补这一差距,并允许高通量酶功能预测。
最近,深度学习,特别是图神经网络(GNNs)变得越来越流行,并被用于各种化学任务,如药物发现、亲和力预测、蛋白质界面预测和蛋白质功能预测。GNNs是结构蛋白质功能预测的流行方法,因为大多数实现都具有等变性和平移不变性。除此之外,图与化学表示密切相关。然而,将蛋白质结构表示为3D图是困难的,且耗费内存和时间。为了避开这些限制,通常构建的图仅包含从3D到线性空间投影的信息。GNNs的最新发展使作者能够在消息传递网络中编码原子之间的位置、距离和角度,并在小分子上进行测试。
在这里,蛋白质功能与酶催化的特定反应相关,这由酶委员会(EC)编号描述。EC编号由四个分层数字表示,分别指定主类、亚类、次亚类功能,以及特定酶功能名称。例如,受体蛋白-酪氨酸激酶(receptor protein-tyrosine kinase),EC编号为2.7.10.1,是一种转移酶(2),转移含磷基团(2.7),作为蛋白质-酪氨酸激酶(2.7.10)。
作者的目的是学习如何利用分子识别特性从结构中预测EC编号。虽然使用机器学习预测EC编号并不新颖,但明确编码3D信息来预测EC编号的基于结构的方法相对较少。
模型部分
作者提出TopEC,一个使用图神经网络进行酶功能预测的软件包。(图1a)。
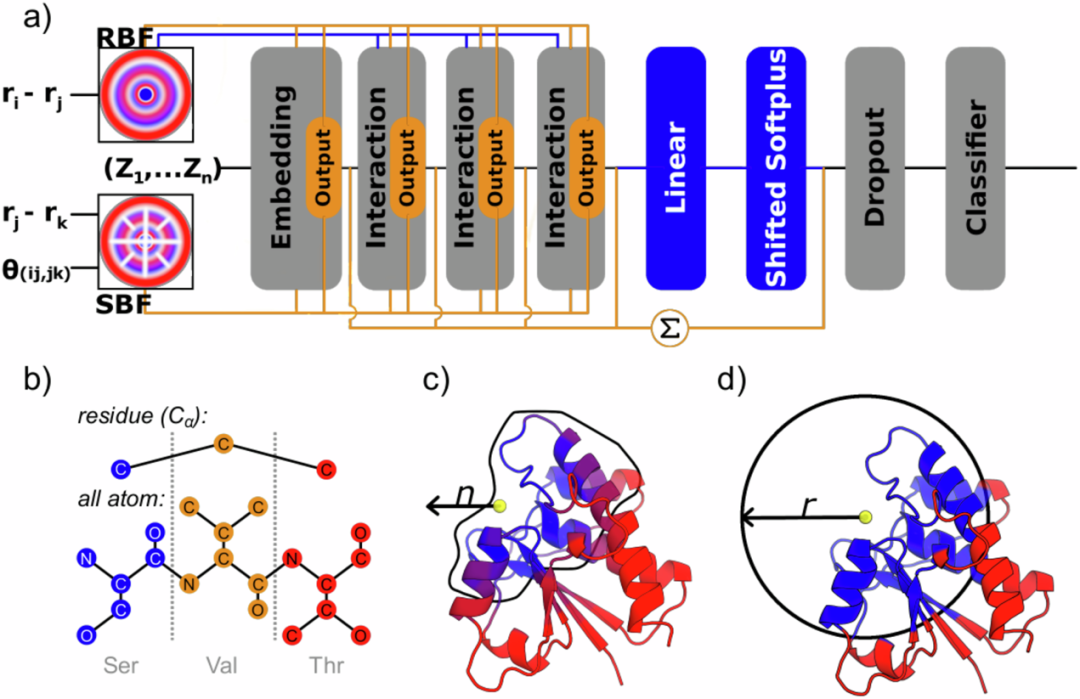
图 1
基于结构信息预测EC编号而不受结构折叠偏差的影响是具有挑战性的。虽然酶的折叠通常决定功能,但即使是微小的突变也能极大地改变功能。例如,TIM桶状结构和Rossman折叠是具有相似超二级结构的组,但催化许多不同的反应。如果作者仅考虑整体形状(折叠),将忽略导致不同功能的微小差异。作者称这为折叠偏差。通常,通过按30%序列相似性对训练、验证和测试分割进行聚类来消除折叠偏差。因此,作者称作者的分割为"折叠分割"。当从训练数据集中移除折叠偏差时,深度神经网络方法往往表现不佳(F score:0.3-0.4)。
为了解决计算需求和折叠偏差的问题,作者引入了基于酶结合位点的局部化3D描述符。为了确定化学转化可能发生的区域,作者使用实验证据、同源注释和预测方法P2Rank。基于这个区域,作者以最近的n个原子(图1c)或定义半径r内的所有原子(图1d)为基础进行酶功能分类。作者比较了两种酶结构分辨率的EC预测(图1b)。在残基分辨率上,作者为酶骨架的每个Cα原子位置创建一个图节点。在原子分辨率上,作者为蛋白质的每个重原子位置创建一个图节点。区域表示相对于完整结构表示有两个优势:(1)作者将网络注意力集中在从蛋白质的结合位点学习酶功能上。(2)作者减少了GPU内存占用,因为单个酶的原子图不适合NVIDIA A100 40 Gb GPU。作者使用区域表示为3D感知消息传递网络SchNet和DimeNet++(图1a)创建TopEC-distances和TopEC-distances + angles。TopEC-distances基于SchNet,用于创建原子和残基分辨率的局部化3D描述符。TopEC-distances + angles基于DimeNet++,用于创建残基分辨率的局部化3D描述符。由于GPU内存限制,作者在原子分辨率上没有获得满意的TopEC-distances + angles结果。
为了评估功能预测性能,作者使用蛋白质中心F score(F1),如DeepFRI中类似任务所用。调和F score通过精确度和召回率的调和平均值测量准确性。整体性能是在测试集中所有酶的平均值,与DeepFRI一样。所有统计数据都使用PyCM包计算。虽然作者在下面显示了详细的分解,但在图中作者仅报告了网络、大小和分辨率参数组合中的最高性能者。每种测试组合的完整统计报告和混淆矩阵可在附录数据1中获得。
使用的数据集
由于PDB中存在高EC冗余和具有实验已知结合位点的多样酶功能数量较少,作者在实验和预测蛋白质结构的数据集组合上训练了网络。作者现在描述包含在作者早先发布的TopEnzyme数据库中的数据集(表1)。
首先,作者使用了实验结构。为此,作者筛选了带有相关EC和结合界面的酶结构的Binding MOAD。这产生了21,333个实验确定的酶,覆盖1625种不同的酶功能。作者称这个筛选集为Binding MOAD数据集。为了补充它,作者用TopModel生成了同源模型。这产生了8904个预测酶结构,覆盖2416种酶功能。这是初始TopEnzyme数据集。作者选择这种方法是因为它提供了来自Binding MOAD的实验确定结合位点和从用于结构预测的同源模板推断出的准确派生结合位点。作者在Binding MOAD和TopEnzyme上测试了两种数据分割:时间分割和如ECPred中的折叠分割。对于折叠分割,作者使用MMseqs2以30%序列同一性对数据库进行聚类。所有数据集都按照大约80%/10%/10%的比例分为训练集、验证集和测试集。作者还测试了Binding MOAD和TopEnzyme的组合,并称之为Combined数据集。
为了获得更大和更多样化的结构数据库用于训练,作者使用预测的结合位点信息补充了实验数据集。作者筛选了PDB,只保留使用折叠分割且至少有50个结构的EC名称,总共有300个酶类,涵盖56,058个结构,形成了PDB300数据集。当缺乏任何实验信息时,作者使用P2Rank获取结合位点。同样,作者使用MMseqs2以30%序列同一性对数据库进行聚类,并大约按80%/10%/10%的比例分为训练集、验证集和测试集。
随着AF2数据库(AF2 DB)的发布,作者进一步用AF2端到端方法生成的酶结构补充了计算数据集。为了充分利用预测的酶结构,作者通过整合AF2 DB中可用的AF2结构扩展了TopEnzyme,并将其作为单独的数据库发布。作者筛选了UniprotKB/Swiss-Prot数据库,寻找带有相关EC的蛋白质。在用P2Rank预测结合位点后,作者移除了所有没有预测结合位点的酶,得到201,107个酶,覆盖703个独特的EC,每个EC至少有50个不同的计算结构,形成了AF703数据集。
为了获得对网络模型性能的生物化学见解,作者引入了两个基准数据集。第一个是修改的Price数据集。Price数据集指的是SEED或KEGG数据库中之前出现的基于从序列信息派生的预测的错误注释或不一致注释的实例。第二个,ProSPECCTs数据集分为十个不同的(子)类别(DS1-DS7,子类别:DS1.2,DS5.2,DS6.2),对酶结合位点的各种特性进行基准测试。每个数据集内容的详细信息可在附录表1中获得。在移除作者没有用AF703和PDB300数据集训练的EC后,作者剩下5543个结构。这些类别因不包含至少50个样本而从训练中移除。
最后,作者调整了催化位点图谱(CSA)数据集以及BioLiP中的所有结合残基。同样,作者移除了所有没有用AF703数据集训练的EC。作者没有用PDB300训练的网络测试CSA。由于CSA作为PDB内的数据库存在,在正确将数据分为训练和测试集后,将没有足够的训练样例。这提供了364个具有实验结构的酶,跨越295个EC,作者拥有完整的结合信息。筛选后的Price、ProSPECCTs和CSA数据集在附录数据2中报告。
结合位点的局部信息足以支撑酶分类
最初,作者使用局部化3D距离描述符测试了对EC分类的七个主要酶功能类别的预测性能。作者通过过采样训练网络,以减少对更多人口主类的偏差。为了比较,作者使用各自出版论文中描述的最佳设置,在数据集上重新训练了EnzyNet(一个3D-CNN)和DeepFRI(一个GCNN)。此外,作者还在结合位点周围16 Å半径切断的局部描述符上重新训练了EnzyNet和DeepFRI。结果显示在表2A中。由于EnzyNet为多种模式返回结果,作者在附录图3中显示了完整结果。
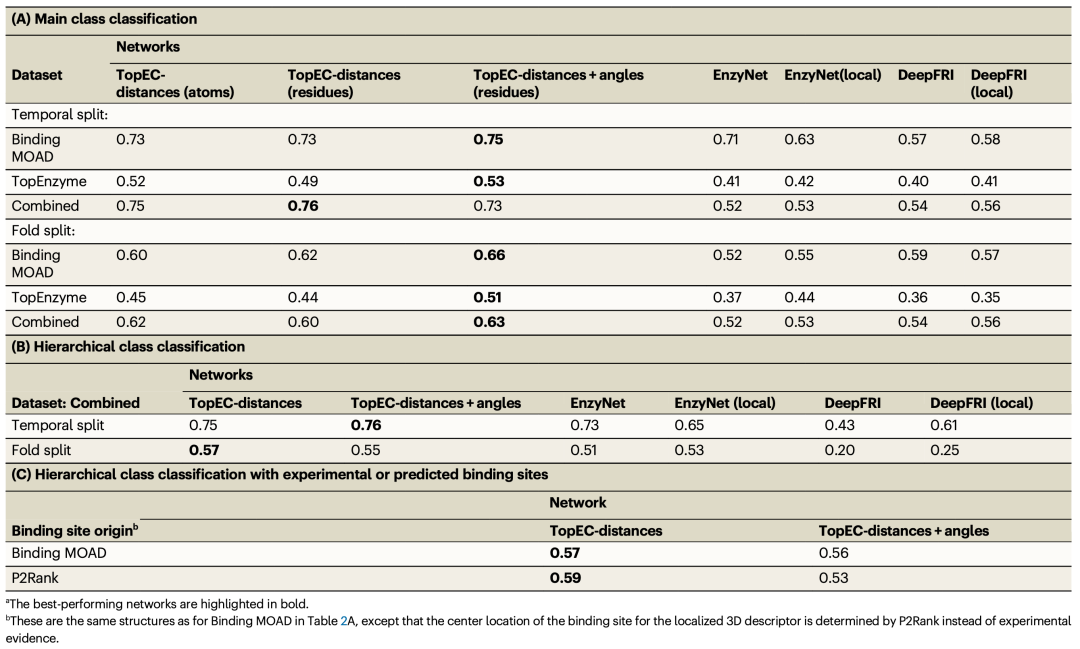
表 2
在六项测试中的五项中,残基分辨率的TopEC-distances + angles优于所有其他神经网络,与TopEC-distances相比F分数平均增加0.04,与EnzyNet和DeepFRI相比更显著地增加0.13。只有在组合数据集的时间分割上,残基分辨率的TopEC-distances优于其他。一般来说,酶的拓扑结构(折叠)被认为是给定功能的主要决定因素。这导致时间分割上的性能更高,因为训练集和测试集中存在许多相同的拓扑结构。在折叠分割的情况下,数据按30%序列同一性分离,在训练、验证或测试集中没有共同的折叠。因此,网络不能从拓扑结构中学习功能,这减少了对具有相似拓扑结构但不同功能的蛋白质的偏差。这种能力对于在酶发现或工程合成酶级联中应用酶功能分类很重要。
TopEnzyme数据集上的性能明显低于Binding MOAD数据集。比较模型的TopEnzyme部分可能不包含足够的样本从训练集中学习局部化学:在这个TopEnzyme部分,对于特定的EC编号,每个测试模型平均有2-3个训练模型。因此,TopEnzyme部分在局部层面上包含各种化学,但每种情况只有少数例子。这可能也是组合数据集上的性能低于Binding MOAD数据集的原因,尽管实验和预测结构的组合已被证明可以改善结果。
在EnzyNet中,酶信息被简化为拓扑视图。它在具有时间分割的Binding MOAD数据集上的表现比在拓扑重叠较少的数据集上更好。在DeepFRI中,酶信息是从2D接触图创建的,一般表现最差。作者的局部描述符对所有数据集的表现都优于EnzyNet和DeepFRI,可能是因为它隐式地将网络引导到与酶功能更相关的区域。只有在时间数据集上,EnzyNet优于局部表示,因为在这里它可以从拓扑视图中学习。
接下来,作者评估了网络在使用组合数据集预测EC编号的完整层次结构时的性能。作者在表2B中展示了TopEC-distances和TopEC-distances + angles与具有局部描述符的DeepFRI和EnzyNet的对比结果。对于时间数据分割,TopEC-distances + angles表现最佳,而对于折叠数据分割,TopEC-distances表现最佳,与EnzyNet和DeepFRI相比,F分数平均增加0.06和0.29。作者还测试了图计数和局部化3D描述符位置对性能的影响。根据模型的不同,性能在每个酶图约75-100个节点时开始趋于平稳。TopEC-distances + angles在更大的图上表现更差,因为作者开始在使用的GPU上遇到内存不足问题。当作者选择随机中心来定义局部化3D描述符的结合位点时,模型表现更差。对于层次分类,与主分类一样,很好地表示局部化学以隐式地将网络引导到相关区域可能比整体拓扑-功能关系更为重要。在其他完整结构方法中,网络的显著性图与实验结合位点重叠。在TopEC中,与EnzyNet和DeepFRI相比,结构信息被明确编码(见上文)。作者的结果表明,在不考虑拓扑-功能关系的情况下分类酶功能时,明确编码结构信息是重要的。
最后,作者使用来自Binding MOAD的实验结合位点或由P2Rank在具有折叠分割的实验结构上预测的结合位点训练了网络(表2C)。虽然结合位点预测器并不完美,但一些结构噪声可能使机器学习方法对不确定信息更加稳健。对于作者的两个网络和两种结合位点来源,预测性能相似。因此,当缺乏实验信息时,作者将使用P2Rank获取更大数据库中的结合位点信息。
加入计算生成的酶结构数据,能够提升网络的预测性能
在测试计算生成的酶结构之前,作者希望获得基线性能。作者使用预测的结合位点为实验确定的酶结构创建了折叠分割,以获得更多训练样本,即PDB300数据集。图2a显示了每个EC的精确率-召回率曲线下面积(AUPR)分布。这个数据集的F分数较低,主类为0.66,亚类为0.53,次亚类为0.45,名称级别为0.39。
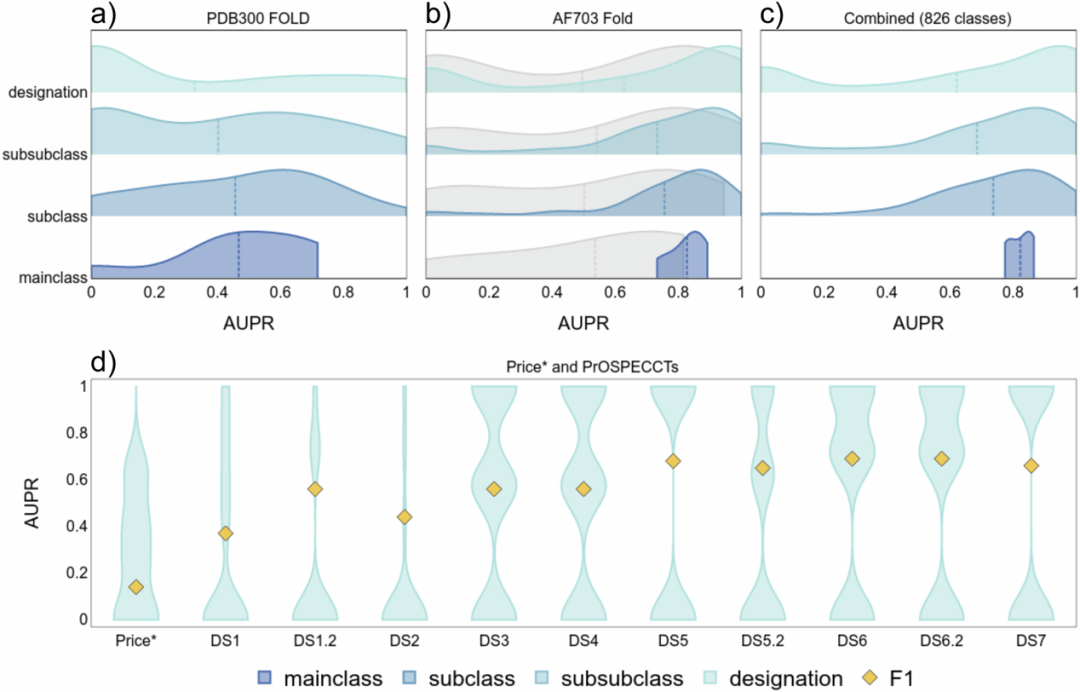
图 2
在用PDB300训练时,作者遇到了一些问题。由于样本有限,许多类别预测效果不佳。例如,对于转位酶(F score:0),作者只有228个用于训练、验证和测试的实验结构。此外,虽然在创建预测模型时实验证据被视为基准事实,但PDB存在已知问题,如冗余或被标记为酶-底物复合物的酶-抑制剂复合物。对于丝氨酸内肽酶La(EC:3.4.21.53),八个测试PDB中没有一个被正确预测。值得注意的是,一个被描述为没有活性(4fwh),三个被描述为与抑制剂复合(4fw9,4fwd,4fwg),其他四个来自他们对催化二联体残基进行突变的突变研究(7ev4,7euy,7ev6,7eux)。然而,这些结构在PDB中都被标记为具有EC分类3.4.21.53的酶活性。
接下来,作者用TopEnzyme的同源建模生成的结构模型或AF2数据库(AF2 DB)中相同Uniprot ID的结构训练了网络。对于在原子分辨率下训练的网络,AF2结构通常提高了预测性能(t检验,n = 7.924:p值 = 0.041)。对于在残基分辨率下训练的网络,性能更为相似。结果表明,比较模型和端到端方法生成的模型在TopEC中表现相当。同源模型的一个优势是作者可以利用晶体结构同源物来优化结合位点预测。
使用AF2结构的折叠分割网络的性能如图2b所示,并与使用实验结构和比较模型组合数据集获得的最佳折叠分割网络进行了比较(表2B)。使用AF2数据集极大地提高了主类(+0.18)、亚类(+0.28)、次亚类(+0.33)和名称(+0.33)级别的F分数。此外,跨越七个主类的名称数量从97增加到703。这导致代表性不足的类别的AUPR显著增加,如裂合酶(+69个新名称,+0.28)、连接酶(+56个新名称,+0.89)和转位酶(+21个新名称,+0.64)。图2b中亚类、次亚类和层次类也获得了类似的向更高AUPR值的分布转移。虽然随着酶功能特异性的降低(即从名称移动到主类),分类结果有所改善,但结构数量、折叠、pLDDT评分或名称与性能之间没有相关性。
在price和ProSPECCT上做基准测试
作者测试了AF2 fold-split网络在修改后的Price和ProSPECCTs数据集上的性能(图2d)。结果显示了EC指定级别的AUPR曲线和叠加的F1分数。虽然Price中酶的平均AUPR值较低,但网络对正确预测(0.87置信度分数)比错误预测(0.56置信度分数)更有信心。有趣的是,对于EC前三位数字正确的情况(0.71置信度分数),置信度分数仍然高于2位或更多位数错误的情况(0.44置信度分数)。这些数据集的设计本身就具有挑战性,即来自其他折叠的这些EC的结构酶数据可用性低,由于训练样本有限导致结果较低。Price数据集中的酶保守性较低,许多酶之前已被体外方法错误分类。与ProteInfer类似,作者尝试恢复新的修正分类。与ProteInfer相比,作者正确预测了更多酶,但也错误预测了更多酶,因为ProteInfer包含了一种不做不确定预测的机制。
一般来说,对于ProSPECCTs,作者要么完全正确地预测酶类,要么完全错误,导致沙漏形状的AUPR曲线。对于错误预测的类别,在移除与ProSPECCTs序列相似性>30%的任何训练样本后,作者缺乏来自不同折叠的EC多样性。
网络从化学作用和局部形状的相互作用中学习
由于作者通过使用折叠分割和局部3D描述符将整体形状与酶功能解耦,网络从局部形状模式中学习。为了仔细研究这一点,作者使用了GNNExplainer的修改版本分析了网络,这是一种用于研究图神经网络的模型无关工具。由于作者已经确定了最能描述酶功能(结合口袋)的子图结构,作者用从GNNExplainer改编的软掩码方法替代子图识别,因为作者只是定性地描述残基或原子的重要性。软掩码方法预期会学习每个节点对预测的相对重要性。首先,作者将GNNExplainer应用于在折叠分割的AlphaFold2结构上训练的TopEC网络,该网络仅使用残基的Cα位置和CSA数据集。因此,作者正确预测了所有四个EC层次的232个功能。正确预测的子集用于GNNExplainer获取节点重要性,该重要性经过归一化并映射到每个残基上,提供了每个残基重要性的概览(图3a)。
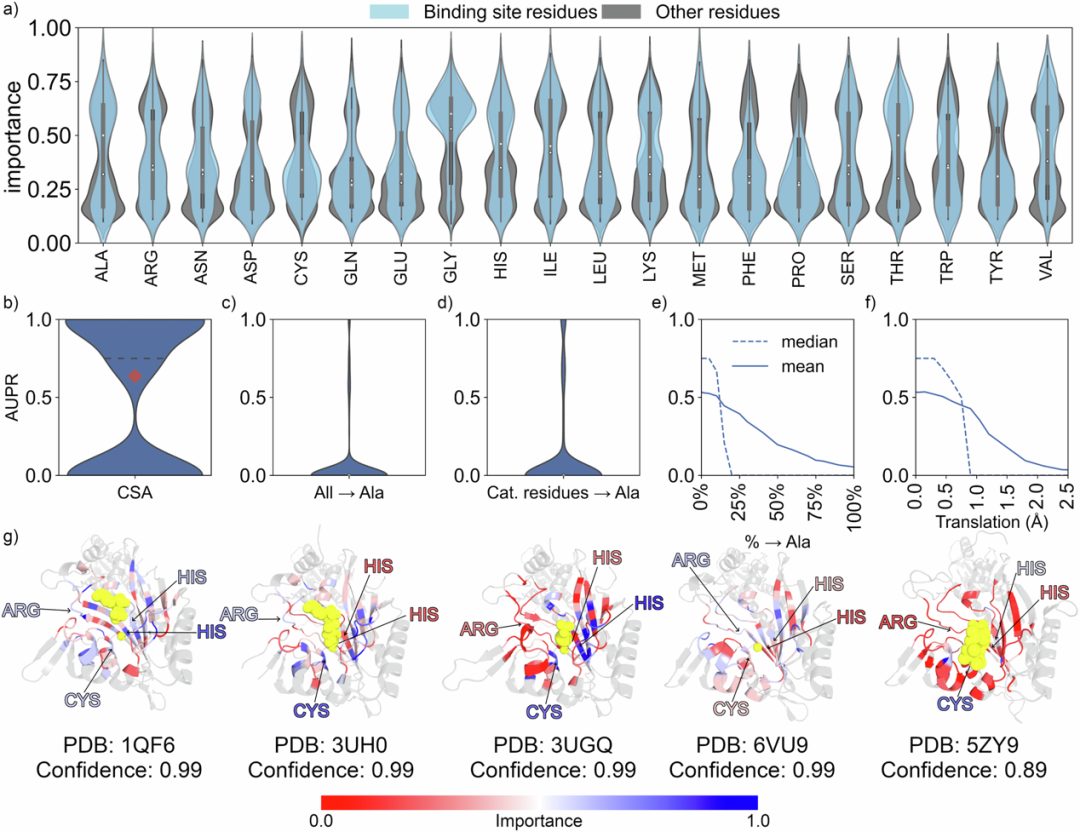
图 3
对于天冬氨酸、甘氨酸和苏氨酸,结合位点和催化残基被确定为更重要。然而,重要残基不仅限于结合位点,如半胱氨酸、苯丙氨酸、脯氨酸和色氨酸所示。甘氨酸是所有四分位数中最重要的残基。由于作者只考虑Cα位置,甘氨酸是网络中化学上描述最完整的残基,而对于其他残基,侧链定位可能至关重要。
为了测试来自局部形状或专家化学知识的化学信息的影响,作者使用了从CSA获得的364个酶。图3b显示了每个类别的AUPR分布。在零和一处分布较广,因为在该数据集中特定类别的酶通常只有一个(364个酶中有295个类别)。为了测试化学与形状的影响,作者计算机模拟将所有残基突变为丙氨酸,这不会改变残基网络的蛋白质形状(图3c)。作者还将仅催化残基突变为丙氨酸(图3d)。在这两种情况下,网络性能都明显下降。作者测试了在失去准确预测所需的化学知识之前,能随机突变多少残基(图3e)。虽然随着更多突变,平均AUPR缓慢下降,但中位数AUPR在突变约20%的残基时急剧下降,表明局部侧链信息对学习过程至关重要。为了测试能扰乱多少形状表示,作者将残基位置随机平移0.0至2.5 Å(图3f)。从0.75 Å开始的平移会降低网络性能,这对应于约4.5 Å57的晶体结构分辨率。
对于来自不同PDB结构的五个正确预测的苏氨酸-tRNA连接酶域,催化残基根据GNNExplainer的归一化重要性进行着色(图3g )。虽然这些结构具有相同的折叠,但这五个结构的序列相似性约为35-60%,除了3UH0和3UGQ之间(100%)。在所有五种情况下,彩色β-折叠片上的残基交替地显示重要性,尽管重要性与侧链朝向底物的方向或不朝向底物的方向无关。重要性差异随着与底物距离的增加而增加,指向了局部化学信息的相关性。
总体而言,破坏局部化学或形状都会降低网络性能。反过来,网络性能可能源于生物化学特征(如特定相互作用或保守基序)、局部形状以及原子类型依赖特征之间的相互作用,这些特征与一般蛋白质生物化学理解更难协调。
编译|黄海涛
审稿|王梓旭
参考资料
van der Weg, K., Merdivan, E., Piraud, M., & Gohlke, H. (2025). TopEC: prediction of Enzyme Commission classes by 3D graph neural networks and localized 3D protein descriptor. Nature Communications, 16(1), 2737.
