
DRUGAI
今天为大家介绍的是来自曼尼托巴大学计算机科学系的Pingzhao Hu教授与微生物学系的Silvia T. Cardona教授团队发表的一篇论文。在早期药物研发中,了解化合物与蛋白质之间的相互作用至关重要,这有助于我们理解分子作用机制并发现潜在的治疗效果。GraphBAN是一个基于图的框架(Graph-based Framework),该框架采用归纳式方法,能够处理未知节点的链接预测,突破了传统方法只能在已知环境下工作的限制。GraphBAN采用教师-学生模型进行知识蒸馏,其中教师模块利用网络结构信息,学生模块专注于节点属性。该框架还包含领域适应模块(Domain Adaptation Module),提高了跨数据集的效果。在5个基准数据集的实验中,GraphBAN的表现超过了10个基线模型,并在Pin1蛋白质的案例研究中证明了其在实际应用中的有效性。

新药研发过程中,了解化合物与蛋白质之间的相互作用至关重要。传统的实验方法往往耗时费力,因此科学家们开发了计算机辅助的预测方法。早期的计算方法主要包括分子对接和分子动力学模拟,这些方法可以预测化合物与蛋白质的结合能力,并模拟它们之间的动态互动。但这些方法存在明显短板:不仅需要强大的计算资源,还依赖高质量的分子结构数据,而这些数据并不总是现成可用的。随着人工智能技术的发展,机器学习和深度学习为解决这一难题提供了新思路。这些方法主要在两个方面实现了突破:首先是数据处理方式的创新,可以同时处理网络结构数据(如化合物-蛋白质相互作用网络)和表格数据;其次是提高了模型的泛化能力,使其不仅能处理与训练数据相似的情况(域内分析),还能应对全新的、差异较大的数据(跨域分析)。
针对现有方法的不足,研究团队开发了GraphBAN 模型。GraphBAN 模型采用知识蒸馏(Knowledge Distillation)架构,包含教师和学生两个模块。教师模块使用图自编码器(Graph Autoencoder, GAE)处理网络属性,学生模块则通过知识蒸馏损失函数学习化合物和蛋白质的初始特征。该模型整合了大语言模型(LLM)和卷积神经网络(CNN)进行特征表示,并使用双线性注意力网络(Bilinear Attention Network, BAN)模块和跨域适应模块来提高预测准确性。
模型架构
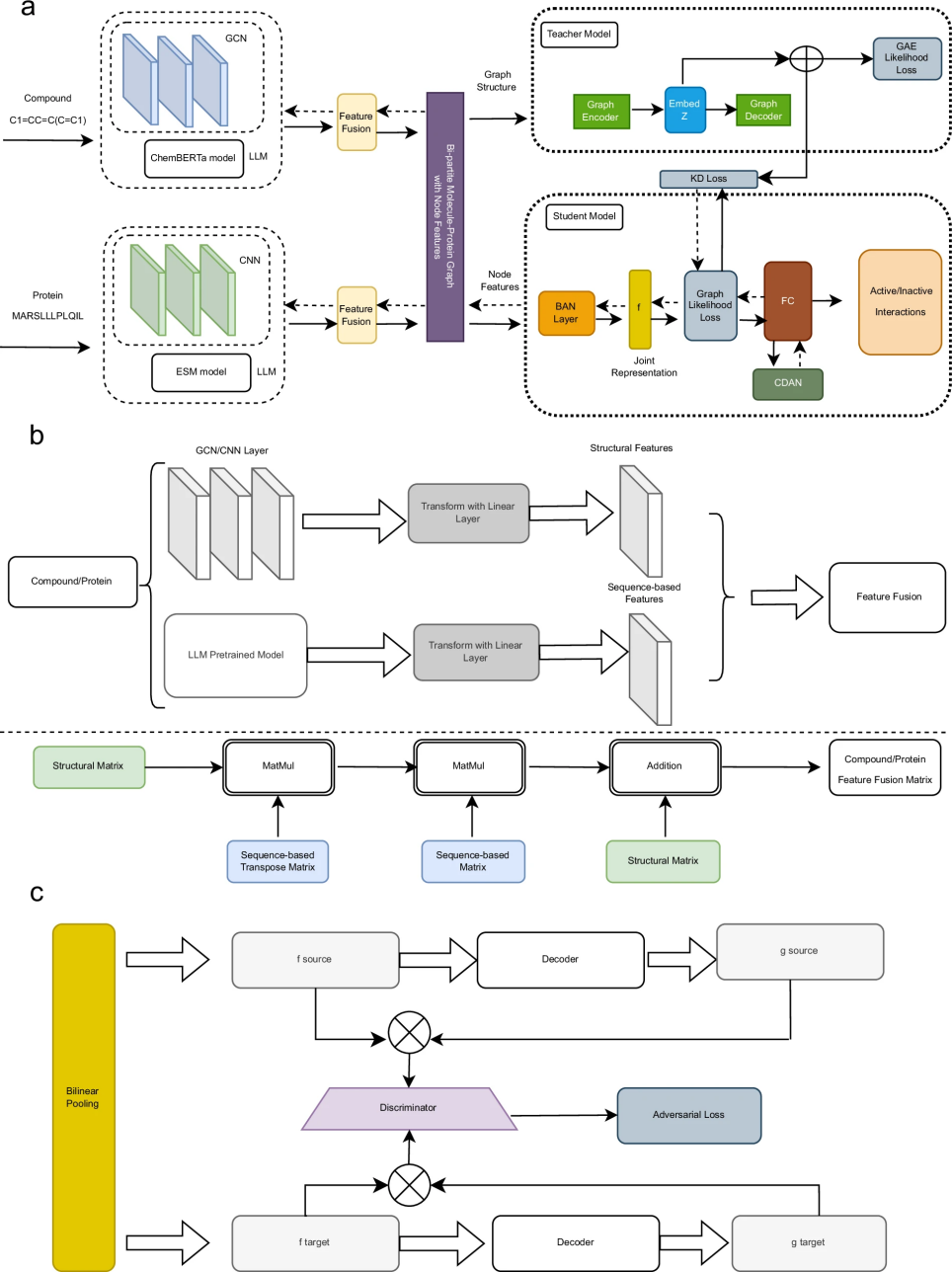
图 1
GraphBAN是一个创新的人工智能框架,其核心架构如图1a所示。该框架能够分析化合物和蛋白质的特征,预测它们之间的相互作用。具体来说,它首先将化合物的分子结构信息和蛋白质的氨基酸序列转换为一个特殊的网络结构,在这个网络中,化合物和蛋白质被表示为节点,它们之间的相互作用则用连接线表示。如图1b所示,该框架采用了多重特征提取方法:对于化合物,它既利用图神经网络分析分子结构,又使用专门的化学语言模型(ChemBERTa)理解分子特征;对于蛋白质,则结合了深度学习(CNN)和进化尺度模型(ESM)来提取特征。此外,如图1c所示,框架还包含了一个特殊的域适应模块,这使得模型能够更好地处理不同类型的数据。
为了验证模型的实用性,研究团队在五个权威数据集上进行了全面测试。这些数据集包含了大量已知的化合物-蛋白质相互作用信息。测试采用了特殊的评估策略:将数据分为两部分,60%用于模型训练(称为源域),40%用于测试模型对新数据的预测能力(称为目标域)。通过多项专业指标的评估,包括预测准确率和召回率等,确保了评估结果的可靠性。每个实验重复进行了5次,以确保结果的稳定性。
实验结果
研究团队首先在多个公开数据集上测试了GraphBAN的性能。GraphBAN在所有测试数据集上都取得了优异成绩,显著超越了现有的顶尖模型(见原文表1)。特别是在BioSNAP数据集上,模型的预测准确率比此前最好的模型提高了将近10%。有趣的是,研究发现模型在不同数据集上的表现存在差异。例如,在规模较小的C.elegans数据集上,模型表现特别出色,这可能是因为小型数据集中的模式更容易识别。而在KIBA数据集上,由于活性和非活性样本数量差异较大,模型的某些评估指标相对较低。
表 2

为了深入理解GraphBAN的成功原因,研究团队进行了详细的模块分析实验(如表2所示)。他们通过逐个移除或添加模型的关键组件,观察每个部分对整体性能的贡献。实验结果令人振奋:特征融合模块能提升预测准确率约5%,注意力网络和域适应模块的组合可以带来接近10%的提升,而教师模块的作用更是显著,能够提高6-9%的性能。这些结果证明,GraphBAN的每个组件都发挥着重要作用,它们的协同效应使得模型能够更准确地预测化合物和蛋白质之间的相互作用。
对于Pin1蛋白质的案例研究
为了验证GraphBAN在实际药物研发中的应用价值,研究团队选择了一个重要的研究目标:Pin1蛋白质。这种蛋白质在细胞周期调节等多个重要生物过程中发挥关键作用,特别是在多种癌症的发展中起着重要作用,因此它是癌症治疗药物开发的重要靶点。
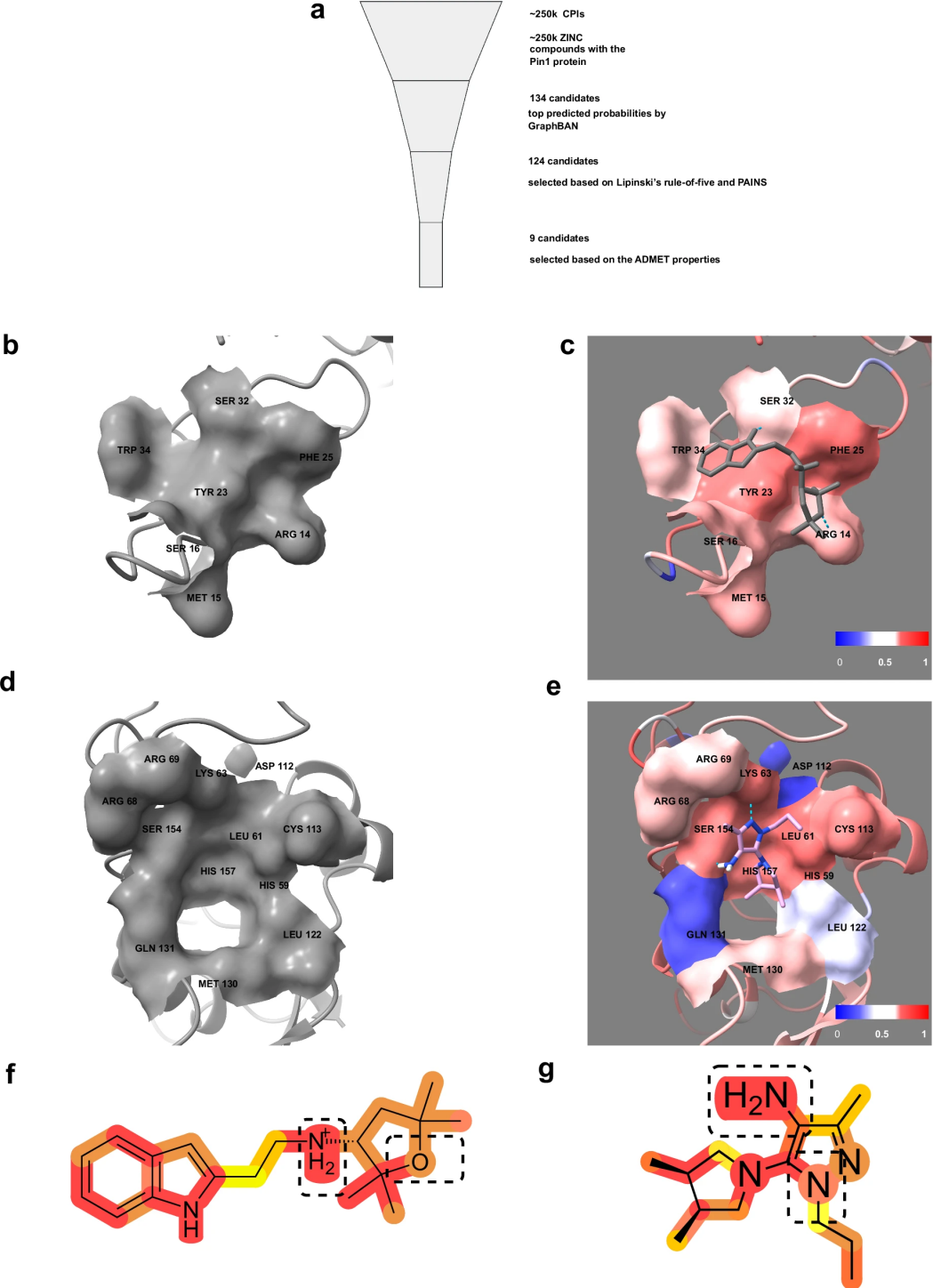
图 2
研究团队使用GraphBAN模型对约25万个候选化合物进行了大规模筛选。如图2所示,筛选过程分为多个步骤:首先,模型从所有候选化合物中识别出134个可能与Pin1发生相互作用的化合物。接着,研究人员通过多重筛选标准进一步缩小范围:首先排除了10个具有不良化学性质的化合物,然后使用人工智能平台ADMET-AI评估剩余化合物的药物特性,包括其在人体内的吸收、分布、代谢、排泄和毒性。最终,研究确定了9个最具潜力的候选化合物。在这9个候选化合物中,研究团队重点分析了两个药物性最好的化合物。如图2b-e所示,这两个化合物都能很好地与Pin1蛋白质结合。更重要的是,当研究人员将这两个化合物与已知的Pin1抑制剂进行比较时,发现它们具有相似的化学特征,这意味着这些新发现的化合物可能具有类似的治疗效果。这一发现不仅证实了GraphBAN模型的可靠性,也为开发新的抗癌药物提供了有价值的候选物。
讨论
文章讨论了GraphBAN模型在药物发现领域的应用和挑战。尽管面临数据集多样性和生物变异性等挑战,但通过知识蒸馏、双线性注意力网络和条件域对抗网络等创新模块,该模型展现出优异的预测能力。在Pin1蛋白质的案例研究中,模型成功从25万个候选化合物中筛选出潜在的抑制剂。未来,通过整合增量学习算法和多模态数据,模型的性能有望进一步提升。
编译|于洲
审稿|王梓旭
参考资料
Hadipour H, Li Y Y, Sun Y, et al. GraphBAN: An inductive graph-based approach for enhanced prediction of compound-protein interactions[J]. Nature Communications, 2025, 16(1): 2541.
内容中包含的图片若涉及版权问题,请及时与我们联系删除
