
DRUGAI
当前的 AI 辅助蛋白质设计主要依赖蛋白质序列和结构信息。然而,大量由人类整理的文本中蕴含了蛋白质功能的高层次知识,其在蛋白质设计中的应用尚未被探索。为弥补这一空白,研究人员提出了 ProteinDT——一个利用文本描述进行蛋白质设计的多模态框架。该框架包括三个步骤:ProteinCLAP(对齐文本与蛋白的表示)、表示生成器(由文本生成蛋白表示)以及序列解码器(将表示转化为蛋白序列)。研究人员构建了包含 44.1 万组文本-蛋白对的大规模数据集 SwissProtCLAP,并在三项挑战性任务中验证了 ProteinDT 的有效性:(1) 文本生成蛋白的准确率超过 90%;(2) 在 12 个零样本编辑任务中表现最佳;(3) 在 6 项蛋白性质预测中有 4 项取得领先表现。
近年来,机器学习在蛋白质发现中展现出巨大潜力,已被广泛应用于蛋白质工程、结构预测、结构重建和反向折叠等任务中。现有方法多聚焦于蛋白质序列和结构的内在属性建模。然而,在小分子药物发现中,结合结构信息与文本描述的多模态方法已取得显著成效,这也引发了一个问题:在蛋白质设计中引入文本描述是否同样有效?
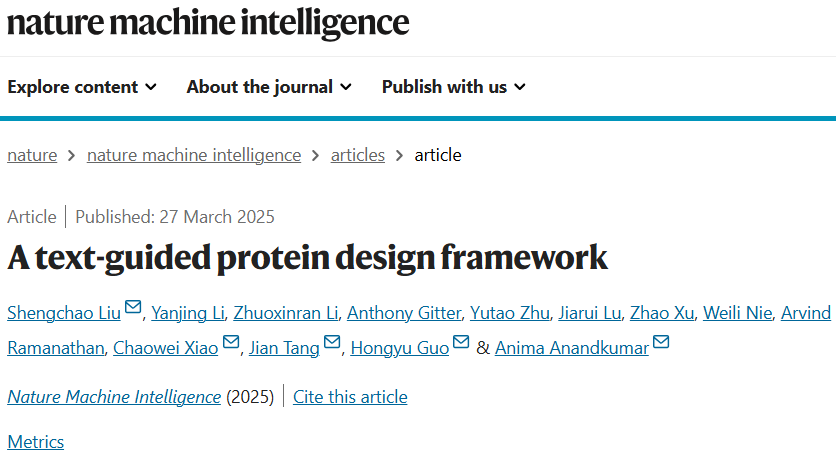
为此,研究人员提出了ProteinDT,一个结合蛋白序列和文本描述的多模态蛋白质设计框架。ProteinDT 利用两个模态:蛋白质序列(反映其结构与功能)和文本描述(如 UniProt 中记录的蛋白功能、定位、参与过程等高阶知识)。两者分别承载生化组成信息与专家总结的知识,有望推动如零样本泛化等更具挑战性的设计任务。
具体而言,ProteinDT 包括三个关键步骤:
ProteinCLAP 模块:通过对比学习对齐蛋白序列与文本描述的表示空间,使用 SwissProtCLAP 数据集进行预训练,包含约 44.1 万组文本-蛋白对。
ProteinFacilitator 模块:从文本模态生成蛋白表示,采用高斯分布建模条件表示。
蛋白序列解码器:基于上述表示生成蛋白序列,结合自回归模型与扩散模型(ProteinDiff)提升生成质量。
研究人员验证了 ProteinDT 在三项任务中的有效性:
文本生成蛋白:从文本提示生成蛋白序列,准确率超过 90%;
零样本蛋白编辑:在 12 项编辑任务中(涵盖结构、稳定性和肽结合等),整体命中率表现最佳;
蛋白性质预测:相比六种主流蛋白表示方法,ProteinDT 在四项基准任务中表现领先。
结果解析
数据集构建与双模态建模
研究人员基于 UniProt 构建了 SwissProtCLAP 数据集,包含 44.1 万对文本-蛋白序列对,涵盖 32.7 万基因和 1.3 万种生物。蛋白质模态采用 ProtBERT 获取序列表示,文本模态则使用 SciBERT 获取功能描述的语义表示。通过预训练,ProteinCLAP 实现了文本与蛋白序列表示的对齐,为多模态理解打下基础。
下游任务一:文本生成蛋白序列
ProteinDT 支持从文本提示直接生成蛋白质序列。研究人员设计了两种策略:
无 Facilitator:直接使用文本表示生成序列;
有 Facilitator:通过 ProteinFacilitator 生成蛋白表示,再生成序列。
实验表明,有 Facilitator 模块显著提升性能,AR-Transformer 解码器表现最好,准确率超过 90%。相比基线模型(如 Galactica、ChatGPT),ProteinDT 展现出更强的生成能力。
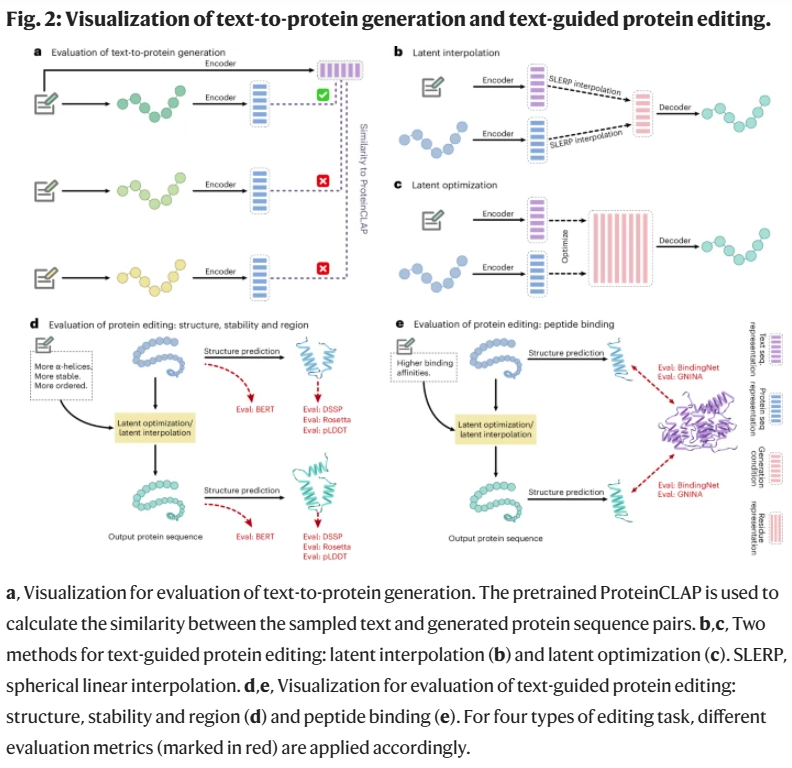
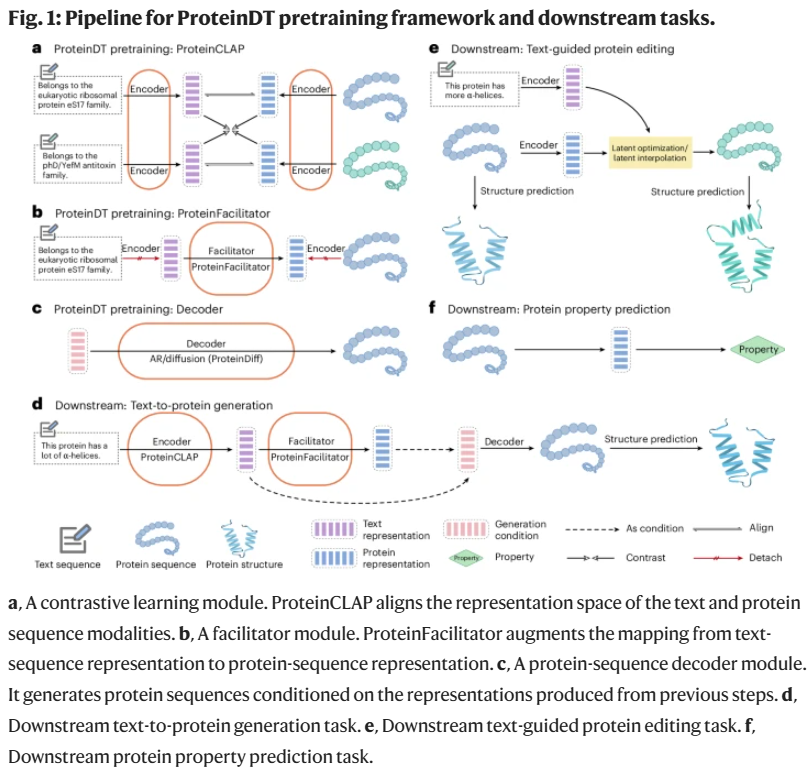
下游任务二:零样本蛋白编辑
ProteinDT 可在无额外训练的情况下,根据文本描述编辑给定蛋白序列。研究人员提出两种编辑方法:
潜表示插值:在文本和原始蛋白表示之间插值生成新表示;
潜表示优化:优化一个同时接近文本和蛋白表示的中间表示。
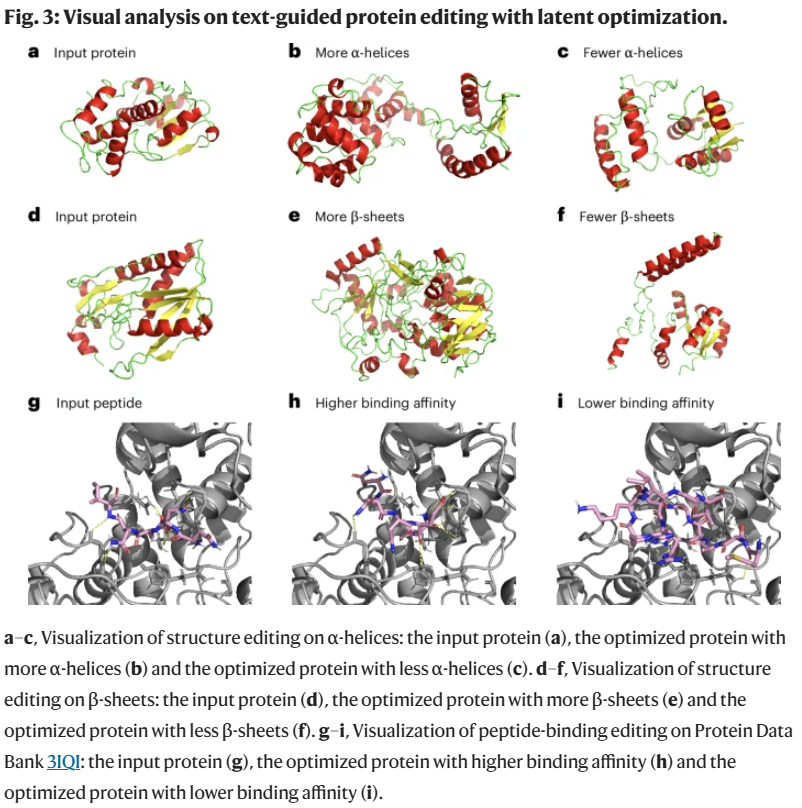
共设计了 12 项编辑任务,涵盖结构编辑、区域编辑、稳定性编辑和肽结合编辑等。结果显示,潜表示优化在多数任务中表现更优,尤其在稳定性任务中效果更为平衡。图示还展示了蛋白结构变化和结合能力的改善,进一步验证方法有效性。
下游任务三:蛋白性质预测
为评估预训练得到的蛋白表示,研究人员在 6 个标准任务上进行了测试,包括二级结构预测、同源预测、接触预测、荧光强度与稳定性预测。
结果显示,ProteinCLAP 表示在结构相关任务中表现最佳,在同源性和稳定性任务中也稳居前三,仅在荧光任务中略逊一筹,显示其在蛋白功能建模上的鲁棒性与泛化能力。
讨论
数据集质量挑战
本研究构建的文本-蛋白配对数据集 ProteinDT 共包含 44.1 万组数据,但相较于图文等领域,这一规模仍较小。若进一步引入实验解析的蛋白骨架结构,数据量将锐减至约 4.5 万对,成为该方向发展的瓶颈。
为缓解数据不足,研究人员采用了预训练模型作为解决方案。但未来也可探索如从生物医学论文中提取蛋白实体,通过命名实体识别与序列映射构建新型数据集。这种方法有望生成规模更大、内容更丰富的数据,但可能会牺牲文本质量与蛋白种类的多样性。
评估方式挑战
尽管文本提示具有高度灵活性,但生成的蛋白仍需进行功能验证。实验手段虽精确,但耗时且成本高。研究中,研究人员借助回归模型作为“代理评估器”模拟实验测试,这是当前强化学习任务中常用的策略。
然而,代理模型的准确性并不总是可靠。为增强评估的可信度,研究人员还引入了结构预测工具 ESMFold 和对接评估工具 GNINA。尽管初步结果令人鼓舞,未来仍需更多维度的验证来全面评估 ProteinDT 在不同编辑任务中的表现。
未来展望:零样本蛋白工程的可能性
ProteinDT 的最终愿景,是在无需实验数据的情况下实现零样本蛋白工程。当前的深度学习方法虽能在特定蛋白功能上表现优异,但仍依赖高通量实验数据。传统方法即便能减少数据需求,也仍需要一定的功能特异性数据支持。
而 ProteinDT 提供了另一种可能 —— 通过挖掘文本描述中蕴含的丰富知识,构建跨模态模型,辅助蛋白质功能设计。这为未来无需实验数据、仅靠文本描述即可定制功能蛋白的设计路径奠定了基础。
整理 | WJM
参考资料
Liu, S., Li, Y., Li, Z. et al. A text-guided protein design framework. Nat Mach Intell (2025).
https://doi.org/10.1038/s42256-025-01011-z
内容中包含的图片若涉及版权问题,请及时与我们联系删除
