

发现新分子(molecules)使人类能够解决健康、农业、能源等领域的问题。分子发现的关键挑战在于,所有可能分子的空间远大于我们能够通过有限资源进行实验测试的分子数量。面对这一挑战,最好的选择是根据我们当前的知识和每次测试可能获得的信息预期,明智地选择要测试的分子。
在机器学习中,这种方法通常称为贝叶斯优化,并且已被应用于许多其他问题,如调优机器学习模型的超参数。尽管理论上贝叶斯优化可以直接应用于新分子的发现问题,但分子的离散特性意味着需要新的模型和算法,才能使贝叶斯优化在实践中发挥作用。
本论文介绍了几种概率机器学习算法,可用于贝叶斯优化循环中,以发现新分子。带权重重新训练的潜在空间优化(第3章)和基于隐函数定理的自适应深度核拟合(deep kernel fitting)(第4章)是两种使用高斯过程与深度神经网络核函数相结合的算法,用于建模分子结构与某些感兴趣性质之间的关系。Tanimoto随机特征(第5章)则允许将一种已建立的化学信息学模型(近似)应用于大规模数据集。最后,逆合成回退(retro-fallback)(第6章)采用了一种新的概率形式化的逆合成问题,来估计一个分子是否可以合成,从而决定它是否应该被贝叶斯优化考虑。
这些算法共同构成了一套工具,可以用来自动化和智能地发现新分子。

作者:Tripp A.
类型:2024年博士论文
学校:University of Cambridge(英国剑桥大学)
下载链接:
链接: https://pan.baidu.com/s/1q6etAfkbxaGKr9roBThCqQ?pwd=fbz9
硕博论文汇总:
链接: https://pan.baidu.com/s/1Gv3R58pgUfHPu4PYFhCSJw?pwd=svp5
引言
尽管时间、资源和聪明才智有限,人类还是发现了具有以下性质的分子:属性让我们能够对其环境进行非凡的控制。青霉素将细菌感染从一种致命疾病变成了一种不便。DDT 等杀虫剂已经从整个大陆消灭了疟疾。避孕药让女性对自己的生殖有了前所未有的控制权。乐观地说,尚未发现的分子将治愈癌症,使全球摆脱化石燃料,或减缓人类衰老。
希望这些例子能够令人信服地证明分子发现是一项值得的努力。从历史上看,许多重要的分子都是偶然发现的——本质上是偶然发现的。1 虽然这种偶然发现在未来无疑会继续存在,但有意识地、系统地发现分子的方法显然是可取的。现在我们考虑如何实现这一点,从一些定义开始。
1.1 “分子发现”的定义和挑战
1.1.1 什么是分子?
虽然这个问题可能看起来过于迂腐,但有许多有用的物质,如金属合金,通常不被认为是“分子”。因此,分子的定义有助于理解本论文的范围。Zumdahl 的经典化学教科书(Zumdahl and Zumdahl,2006,第 52 页)对分子进行了以下解释:
将化合物中的原子结合在一起的力称为化学键。原子形成键的一种方式是通过共享电子。这些键称为共价键,由此产生的原子集合称为分子。
因此,在本文中,我们将简单地将分子视为由键连接的原子的集合。2 这个定义当然是不完美的,但不可否认的是,它包括了大多数小分子药物和肽,这是本文算法的主要设想应用。最终,这个定义促使将分子表示为数学图形(§2.1)。
1.1.2 什么是“发现”?
同样,这个问题可能看起来很迂腐,但究竟什么是“发现”却没有看上去那么清楚。例如,有些分子的特性是古代文明所熟知和利用的,但在过去几个世纪才被化学鉴定出来(例如许多植物染料)。另一种值得考虑的情况是,如果某种分子被“预测”存在或具有某些特性,但直到很久以后才被合成。在这两种情况下,什么应该被视为“发现”尚不清楚。
在本论文中,我们将采用务实的定义,即发现既是分子的识别,也是某些感兴趣属性的测量。特别是,由于此定义要求既知道分子又知道属性,因此将两者之间的关系描述为数学函数是有意义的,从而使分子发现以后可以形式化为优化(§2.2)。
1.1.3 是什么让分子发现变得如此具有挑战性?
发现分子本质上是一个“大海捞针”的问题。有许多潜在的分子需要测试(一些估计表明有 1060 个(Bohacek 等人,1996 年)),但通常只有极小一部分分子具有任何特定的特性(例如治愈细菌感染)。这一挑战进一步加剧了这一挑战,因为感兴趣的特性(例如治疗结果)很难准确预测,而且实验测量成本高昂。这限制了在发现过程中可以获得的信息量。
这些因素使得对每个分子进行详尽测试变得不可行,简单的随机测试策略也无效。为了有效地发现分子,必须清楚地了解哪些分子可能具有所需的特性,然后非常仔细地选择要测试的分子,以免“浪费”任何实验。即使拥有大量专业知识和大量研究预算,分子发现活动仍然会经常失败(经常引用“超过 90% 的药物发现项目失败”这一统计数据,例如 Hay 等人 (2014))。
1.2 为什么要考虑概率机器学习?
尽管计算机对化学的了解可能并不比专家多,但它们在处理信息方面更胜一筹,这一点在化学进入“大数据”时代时非常重要。现代高通量筛选方法可以快速产生数千次测量,而“虚拟库”最多可包含 1020 个分子(Hoffmann 和 Gastreich,2019 年):这个数字太大了,即使是最聪明的人也无法完全处理和分析。仅仅能够全面考虑所有数据和后续实验的所有选项,计算机算法就成为分子发现的重要工具。
从历史上看,分子发现中最流行的算法类别是基于物理的模型,包括对接(Pinzi 和 Rastelli,2019 年)、分子动力学(Durrant 和 McCammon,2011 年)和密度泛函理论(Jones,2015 年)。这些方法对科学家来说非常有吸引力,因为它们具有原则性、可解释性和可推广性。然而,在这些方法中,准确性和计算成本之间总是存在权衡,最准确的模拟非常昂贵(通常需要几天或几周的时间才能在大型超级计算机上运行)。即使拥有巨大的计算资源,最准确的方法仍然会做出简化的假设,因此永远不会完全符合现实。最终,基于物理的算法可以被认为是产生从“便宜但非常不准确”到“昂贵但仍然不完美”的预测。可以理解的是,化学家发现这种权衡令人沮丧和不满意。
在这种背景下,放弃第一性原理并简单地“曲线拟合”观测数据成为一种潜在的吸引人的选择。我们将广泛地将所有此类技术称为机器学习。但是,本文中的算法都可以归类为概率机器学习算法:这意味着它们具有对随机变量执行统计推断的解释,例如实验室测量的结果。对某些读者来说,使用随机性可能看起来很奇怪,特别是在化学背景下,大多数感兴趣的数量都是非随机的物理化学性质。然而,随机性是模拟不确定性的有用工具。由于物理化学性质是使用不完善的设备测量的,因此对于已测量的数量总是存在不确定性,当然对于未测量的数量存在更大的不确定性。不确定性估计在实验进行之前对实验结果提供了“信心”,因此对于决策非常有用。
除了不确定性之外,用概率解释来设计机器学习算法为算法开发提供了一个原则框架,而不是简单地将许多步骤临时串联起来。这给概率机器学习算法带来了可解释性,许多科学家都觉得这很有吸引力。
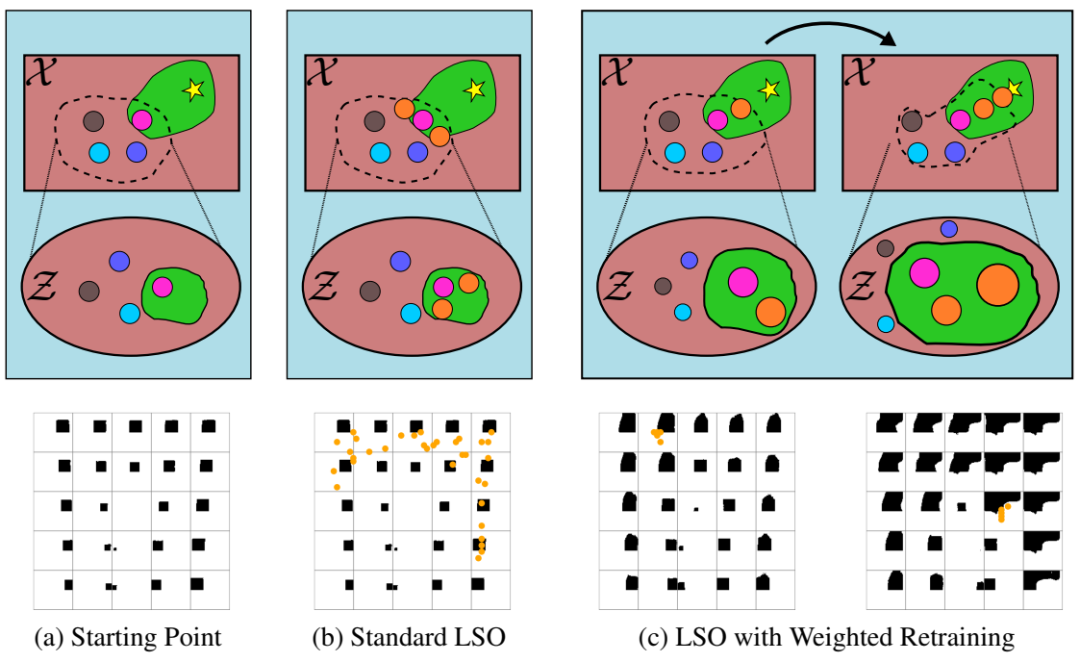
示意图显示了有无加权再训练的 LSO。该卡通说明了生成模型的输入/潜在空间(顶部)。显示了第 3.5.3 节的 2D 形状面积最大化任务中的潜在流形以供比较(底部)。流形中的每个图像都显示了在 2D 潜在空间中解码均匀方形网格上的潜在点的结果;图像以原始网格点为中心。红色/绿色区域分别对应于具有低/高目标函数值的点。黄色星号是 X 中的全局最优值。彩色圆圈是数据点;它们的半径代表它们的权重。虚线围绕由 g 建模的 X 区域(即 g(Z),Z 的图像)。(a)生成模型 g 在优化开始时的状态。(b)g 固定的标准 LSO 的结果,查询橙色点。它只能找到靠近用于学习 Z 的训练数据的点,导致对 X 的探索缓慢且不完整。(c)使用我们提出的方法在 LSO 中途(左)和结束时(右)得到的结果,该方法根据数据点的目标函数值对其进行加权,并重新训练 g 以纳入新查询的数据。这会不断调整 Z 以专注于对 X 中最有希望的区域进行建模,从而加快优化速度并允许在初始训练数据之外进行大量推断。
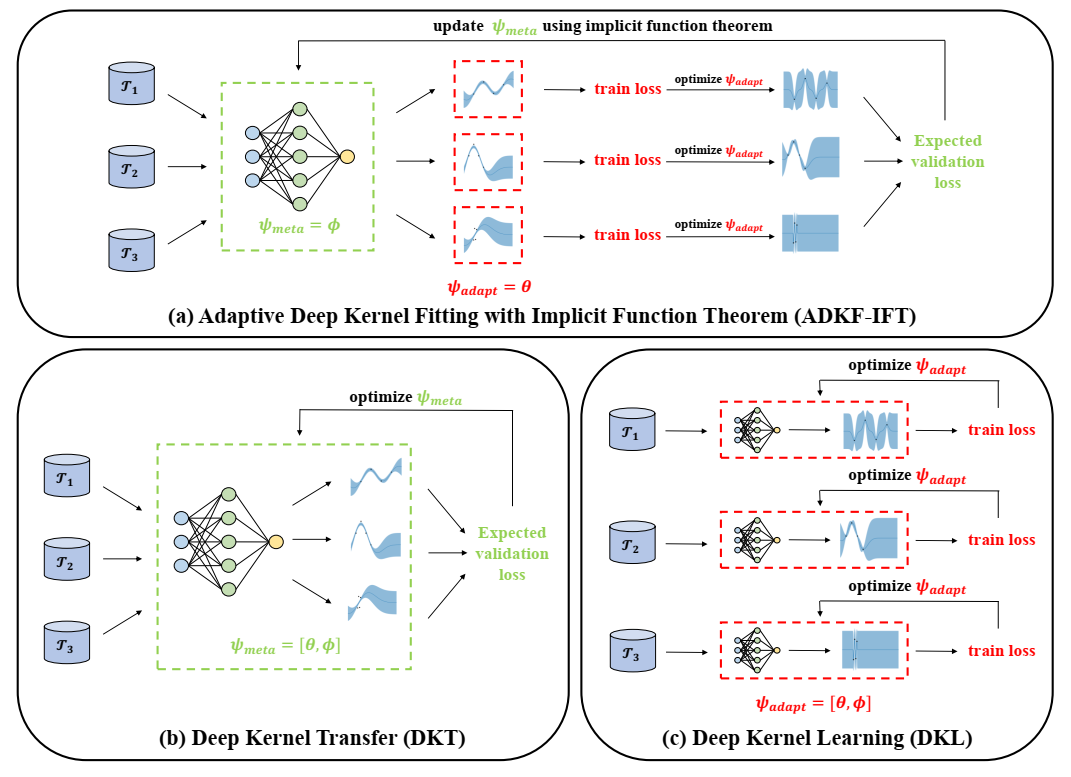
对比图展示了 ADKF-IFT、DKT 和 DKL 的训练过程。 (a) ADKF-IFT 元学习一些参数并调整其他参数。 (b) DKT 元学习所有参数。 (c) DKL 调整所有参数。

当然,retro-fallback 的实际实现可能与算法 6.1 略有不同,它使用缓存和矢量化操作等技术来提高效率。






微信群


内容中包含的图片若涉及版权问题,请及时与我们联系删除
