
At the end of last month, DeepMind, Google’s machine learning research branch known for building bots that beat world champions at Go and StarCraft II, hit a new benchmark: accurately predicting the structure of proteins. If their results are as good as the team claims, their model, AlphaFold, could be a major boon for both drug discovery and fundamental biological research. But how does this new neural-network-based model work? In this post, I’ll try to give you a brief but semi-deep dive behind both the machine learning and biology that power this model.
First, a quick biology primer: The functions of proteins in the body are entirely defined by their three-dimensional structures. For example, it’s the notorious “spike proteins’’ which stud coronavirus that allows the virus to enter our cells. Meanwhile, mRNA vaccines like Moderna’s and Pfizer’s replicate the shape of those spike proteins, causing the body to produce an immune response. But historically, determining protein structures (via experimental techniques like X-ray crystallography, nuclear magnetic resonance, and cryo-electron microscopy) has been difficult, slow, and expensive. Plus, for some types of proteins, these techniques don’t work at all.
In theory, though, the entirety of a protein’s 3D shape should be determined by the string of amino acids that make it up. And we can determine a protein’s amino acid sequences easily, via DNA sequencing (remember from Bio 101 how your DNA codes for amino acid sequences?). But in practice, predicting protein structure from amino acid sequences has been a hair-pullingly difficult task we’ve been trying to solve for decades.
This is where AlphaFold comes in. It’s a neural-network-based algorithm that’s performed astonishingly well on the protein folding problem, so much so that it seems to rival in quality the traditional slow and expensive imaging methods.
Sadly for nerds like me, we can’t know exactly AlphaFold works because the official paper has yet to be published and peer reviewed. Until then, all we have to go off of is the company’s blog post. But since AlphaFold (2) is actually an iteration on a slightly older model (AlphaFold 1) published last year, we can make some pretty good guesses. In this post, I’ll focus on two core pieces: the underlying neural architecture of AlphaFold 2 and how it managed to make effective use of unlabeled data.
First, this new breakthrough is not so different from a similar AI breakthrough I wrote about a few months ago, GPT-3. GPT-3 was a large language model built by OpenAI that could write impressively human-like poems, sonnets, jokes, and even code samples. What made GPT-3 so powerful was that it was trained on a very, very large dataset, and based on a type of neural network called a “Transformer.”
Transformers, invented in 2017, really do seem to be the magic machine learning hammer that cracks open problems in every domain. In an intro machine learning class, you’ll often learn to use different model architectures for different data types: convolutional neural networks are for analyzing images; recurrent neural networks are for analyzing text. Transformers were originally invented to do machine translation, but they appear to be effective much more broadly, able to understand text, images, and, now, proteins. So one of the major differences between AlphaFold 1 and AlphaFold 2 is that the former used concurrent neural networks (CNNs) and the new version uses Transformers.
Now let’s talk about the data that was used to train AlphaFold. According to the blog post, the model was trained on a public dataset of 170,000 proteins with known structures, and a much larger database of protein sequences with unknown structures. The public dataset of known proteins serves as the model’s labeled training dataset, a ground truth. Size is relative, but based on my experience, 170,000 “labeled” examples is a pretty small training dataset for such a complex problem. That says to me the authors must have done a good job of taking advantage of that “unlabeled” dataset of proteins with unknown structures.
But what good is a dataset of protein sequences with mystery shapes? It turns out that figuring out how to learn from unlabeled data–”unsupervised learning”–has enabled lots of recent AI breakthroughs. GPT-3, for example, was trained on a huge corpus of unlabeled text data scraped from the web. Given a slice of a sentence, it had to predict which words came next, a task known as “next word prediction,” which forced it to learn something about the underlying structure of language. The technique has also been adopted to images, too: slice an image in half, and ask a model to predict what the bottom of the image should look like just from the top:

Image from https://openai.com/blog/image-gpt/
The idea is that, if you don’t have enough data to train a model to do what you want, train it to do something similar on a task that you do have enough data for, a task that forces it to learn something about the underlying structure of language, or images, or proteins. Then you can fine-tune it for the task you really wanted it to do.
One extremely popular way to do this is via embeddings. Embeddings are a way of mapping data to vectors whose position in space capture meaning. One famous example is Word2Vec: it’s a tool for taking a word (i.e. “hammer”) and mapping it to n-dimensional space so that similar words (“screw driver,” “nail”) are mapped nearby. And, like GPT-3, it was trained on a dataset of unlabeled text.
So what’s the equivalent of Word2Vec for molecular biology? How do we squeeze knowledge from amino acid chains with unknown, unlabeled structures? One technique is to look at clusters of proteins with similar amino acid sequences. Often, one protein sequence might be similar to another because the two share a similar evolutionary origin. The more similar those amino acid sequences, the more likely those proteins serve a similar purpose for the organisms they’re made in, which means, in turn, they’re more likely to share a similar structure.
So the first step is to determine how similar two amino acid sequences are. To do that, biologists typically compute something called an MSA or Multiple Sequence Alignment. One amino acid sequence may be very similar to another, but it may have some extra or “inserted” amino acids that make it longer than the other. MSA is a way of adding gaps to make the sequences line up as closely as possible.
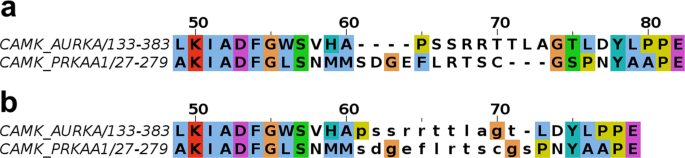
Image of an MSA. Modi, V., Dunbrack, R.L. A Structurally-Validated Multiple Sequence Alignment of 497 Human Protein Kinase Domains. Sci Rep 9, 19790 (2019).
According to the diagram in DeepMind’s blog post, MSA appears to be an important early step in the model.
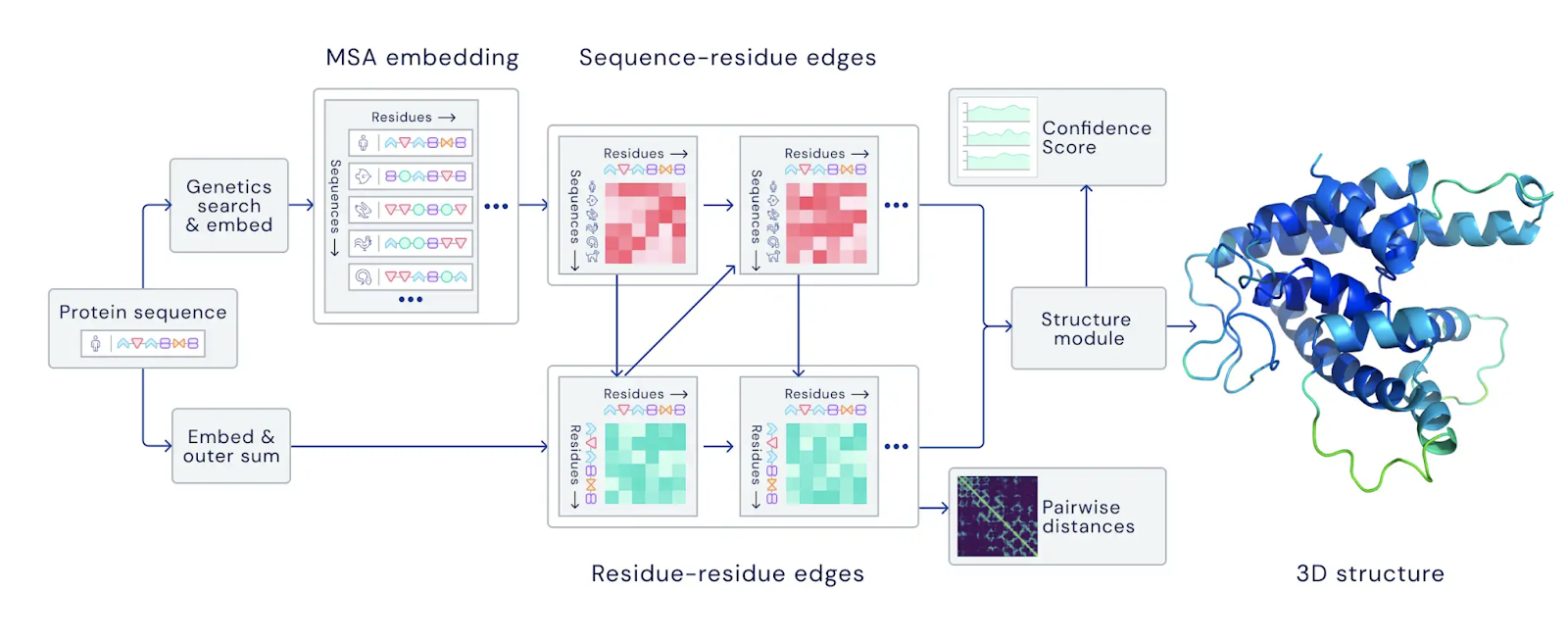
Diagram from the AlphaFold blog post.
You can also see from that diagram that DeepMind is computing an MSA embedding. This is where they’re taking advantage of all of that unlabeled data. To grok this one, I had to call in a favor with my Harvard biologist friend. It turns out that in sets of similar (but not identical) proteins, the ways in which amino acid sequences differ is often correlated. For example, maybe a mutation in the 13th amino acid is often accompanied by a mutation in the 27th. Amino acids that are far apart in a sequence typically shouldn’t have much effect on each other, unless they’re close in 3D space when the protein folds–a valuable hint for predicting the overall shape of a protein. So, even though we don’t know the shapes of the sequences in this unlabeled dataset, these correlated mutations are informative. Neural networks can learn from patterns like these, distilling them as embedding layers, which seems to be what AlphaFold 2 is doing.
And that, in a nutshell, is a primer on some of the machine learning and biology behind AlphaFold 2. Of course, we’ll have to wait until the paper is published to know the full scoop. Here’s hoping it really is as powerful as we think it is.
