

Stanford CS224W: Machine Learning with Graphs
Tutorials of machine learning on graphs using PyG, written by Stanford students in CS224W.
动机
蛋白质(Proteins)是一类生物大分子(biological macromolecules),在获取和储存能量、维持细胞结构完整性以及在细胞或生物体之间传递信息方面发挥着至关重要的作用。这些分子是由线性氨基酸链(linear chains of amino acids)组成的聚合物(polymers),其序列(sequence)决定了蛋白质的结构并最终决定其功能。有 20 种典型氨基酸—所有生命领域所共有的—都具有相同的基本分子框架,仅在侧链功能团(side chain functional groups)方面有所不同(图 1)。利用这些构建块,大自然构建了数百万种不同的结构。对于希望从头或从头构建具有原子级精度(atomic-level accuracy)的功能性蛋白质的蛋白质设计师来说,我们必须能够预测将生成具有所需功能的蛋白质结构的氨基酸序列(amino acid sequences)。鉴于可能的氨基酸序列的组合复杂性巨大(对于长度为 n 的蛋白质,组合复杂性为 20^n),限制搜索空间(search space)的计算方法是必要的。
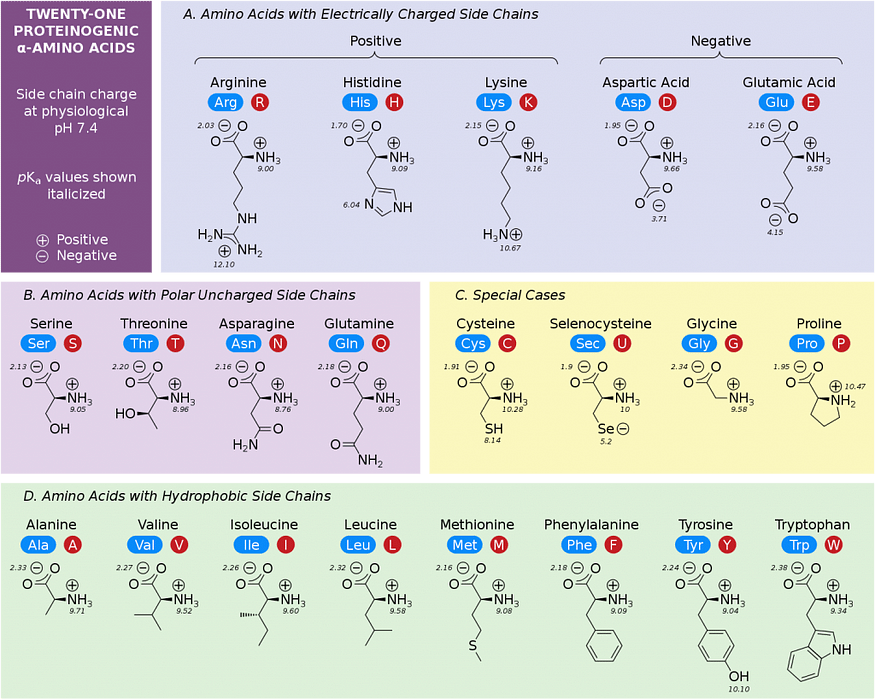
图 1:二十种典型氨基酸。(https://chemistrytalk.org/amino-acid-chart/)
一个挑战是设计能够特异性结合小分子配体的蛋白质。此类蛋白质可能在催化新化学反应(catalyzing novel chemical reactions)、隔离或灭活有害毒素(sequestering or inactivating harmful toxins)以及调节细胞间化学信号(modulating chemical signals between cells)方面发挥重要作用。图 2显示了一种代表性的配体结合蛋白(ligand-binding protein)。目前已有设计配体结合蛋白主链结构的计算方法,但仍需要序列预测算法(sequence prediction algorithms)来根据配体类型预测这些生成结构的氨基酸序列¹。这将是我们项目的重点。
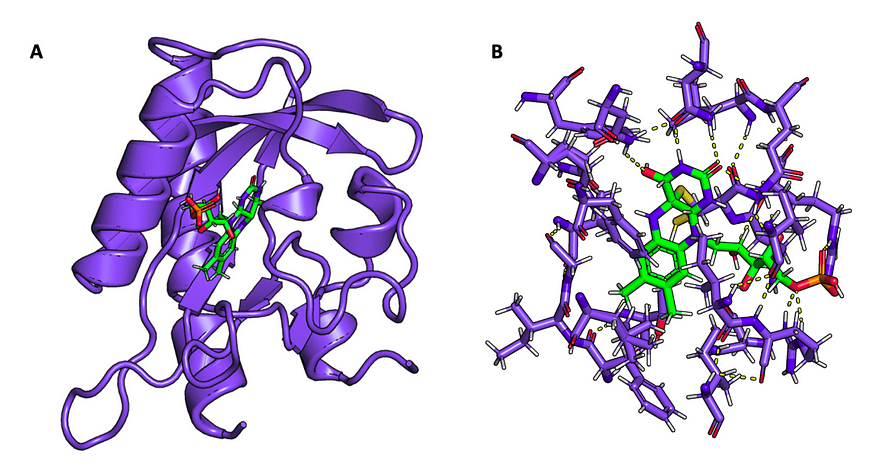
图 2:配体结合蛋白的代表性结构(PDB ID:2V1B)。A ) 燕麦 LOV2(淡紫色)的 N 端和 C 端螺旋以及结合黄素单核苷酸 (FMN) 配体(绿色)。B) 允许 LOV2 以形状和化学互补性结合 FMN 的局部氨基酸环境。有利的相互作用(氢键)显示为虚线黄色线。
任务和数据
在这个项目中,我们实施了一个图神经网络(Graph Neural Network),以解决根据蛋白质结合位点的三维骨架结构和配体的化学结构预测蛋白质配体结合口袋的氨基酸序列的任务。我们将使用 MISATO 数据集²,其中包含来自 PDBbind³ 的 19,433 个蛋白质-配体复合物的结构和序列。为了扩充 PDBbind 数据,Siebenmorgen 等人进行了分子动力学 (molecular dynamic, MD) 模拟以探索这些复合物的构象景观,并进行了量子力学 (quantum mechanical, QM) 计算以提供小分子配体的更丰富的化学数据²。MD 模拟很有用,因为蛋白质结构的静态截图可能无法捕捉动态蛋白质-配体相互作用的真实行为,因此使用 MD 捕获复合物的多种状态可以为序列预测提供更丰富的数据。此外,MD 还消除了 X 射线晶体学(X-ray crystallography)和低温电子显微镜(cryo-electron microscopy)固有的许多伪影,这些伪影可能会降低模型的性能。用户应该能够输入与蛋白质结合口袋复合的化学有效配体,模型将提供配体结合口袋中每种氨基酸的每种氨基酸身份的概率。
#@title Input
import warnings, os, re
from pathlib import Path
from Bio import BiopythonWarning
warnings.simplefilter(action='ignore', category=FutureWarning)
warnings.simplefilter(action='ignore', category=BiopythonWarning)
# pdb='2V1B' #@param {type:"string"}
# ligands="H_FMN" #@param {type:"string"}
pdb='5zxd' #@param {type:"string"}
ligands="H_ATP" #@param {type:"string"}
seed='0' #@param {type:"string"}
noise='0.0' #@param {type:"string"}
knn_res='48' #@param {type:"string"}
knn_atom='25' #@param {type:"string"}
#@markdown * `pdb`: PDB code (from RCSB), or leave blank
#@markdown * `ligands`: comma-separated list of ligands to keep in the structure (name is formatted as H_{name}) with "name" being the three letter ligand name in the PDB file
#@markdown * `seed`: Random seed for the model
#@markdown * `noise`: Standard deviation of Gaussian noise to add to each coordinate
#@markdown * `knn_res`: Number of nearest neighbors where a protein residue node has edges with other protein residue nodes
#@markdown * `knn_atom`: Number of nearest neighbors with which a protein residue node has edges with ligand atom nodes
seed = int(seed)
pdb_path = get_pdb(pdb)
parser = PDBParser()
structure = parser.get_structure("s", pdb_path)
remove_water(structure)
remove_altloc(structure)
remove_hetero(structure, keep_hetero=ligands.split(","))
pdb_path = Path(pdb_path)
pdb_path = f"{pdb_path.stem}_input.pdb"
io = PDBIO()
io.set_structure(structure)
io.save(pdb_path)
print(f"Processed PDB: {pdb_path}")

模型
我们将蛋白质-配体复合物表示为具有两种节点类型的异构图(heterogeneous graph):配体原子(ligand atoms)和残基(residues)。原子节点特征是原子元素类型 (C、N、O、S、P、Zn、Fe、Cu) 的one-hot编码。按照 Dauparas等人的 ProteinMPNN ⁴,残基节点的特征初始化为全零。在我们的模型中,三种关系类型为 (原子,附近,残基) (atom, near, residue)、 (残基,附近,原子)(residue, near, atom)和(残基,附近,残基)(residue, near, residue)。每种关系类型都有自己的一组权重。原子之间的边用两个原子之间的欧几里得距离的离散表示来特征化。残基和原子之间的边用配体原子和四个主链原子 (N、Cα、C、O) 之间的欧几里得距离的离散表示来特征化,每个主链原子一个向量。残基之间的边通过两个残基的四个骨架原子之间的欧几里得距离的离散表示来表征。具体来说,对于每个距离,我们通过将距离传递给径向基函(radial basis function)数来离散化。直观地说,这比标量边权重(scalar edge weights)效果更好,因为距离现在立即嵌入到高维空间中,每个距离范围都会产生不同的高维嵌入。径向基函数使这种分箱变得柔和,并使用多个嵌入的线性组合而不是简单的查找表来产生更丰富的表示。在这些距离中注入了少量高斯噪声(Gaussian noise),以防止过度拟合并解释蛋白质-配体复合物固有的轻微原子扰动(slight atomic perturbations)。选择使用异构图是很自然的,因为我们的数据包含两种不同类型的对象:残基和原子。它们代表不同的概念,残基(residue)/残基、残基/原子(atom)和原子/原子之间的关系是不同的。
该模型的框架采用了具有求和聚合函数的 GENConv 架构(如下所示的公式,xj 是节点 Vj 的特征,eji 是 Vj 和 Vi 之间的边特征)⁵。该框架提供了一种灵活的方式来表示多个节点和关系类型,同时包含边特征。对于这种用例,重要的是用不同的节点和关系类型表示配体原子和氨基酸,因为两个配体原子、两个氨基酸以及配体原子和氨基酸之间的生化关系(biochemical relationship)都是根本不同的,并且图神经网络中节点之间的信息交换应该反映这些差异。此外,边特征至关重要,因为它们编码了有关原子和残基之间距离的信息,这决定了某些相互作用是有利的还是化学无效的,从而告知配体周围的氨基酸概率。我们选择了 1 跳(1-hop)图结构,因为我们推断多跳相互作用不太可能告知氨基酸序列。在配体结合口袋中,附近的配体原子最有可能对氨基酸类型产生最大影响。
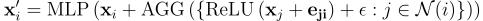
为了进行训练,残基节点嵌入被线性投影到一个 23 维向量中,每个向量代表 20 种氨基酸,一个代表配体的类(残基始终为零),一个掩码类和一个未知类。分类交叉熵损失被用作训练目标。为了进行预测,最终的残基节点嵌入被线性投影到一个 23 维向量中并通过 softmax 函数。最高的 softmax 概率被视为预测的氨基酸身份。
见解和结果
我们发现性能最好的模型是 GINEConv⁶ 和 GENConv⁵ 模型。由于我们将节点特征初始化为全零,因此模型仅基于边信息进行训练。GINEConv 和 GENConv 都通过将节点 Vi 的边特征投影到与 Vi 的嵌入相同的隐藏维度来自然地整合边特征。最终,我们决定使用 GENConv 架构,因为主要区别在于 GINEConv 能够指定任意消息函数而不是 MLP。
训练准确率稳定在 40% 左右,而验证准确率稳定在 30% 左右(图 3)。虽然这些准确率看起来很低,但相比之下,以结构为条件的序列设计的基础计算方法是 Rosetta,其准确率约为 32%。虽然对序列设计模型进行基准测试的最常见指标是准确率(也称为序列恢复率),但我们注意到,高性能模型不应具有极高的序列恢复率(例如超过 80%)。蛋白质序列具有天然冗余,其中具有相似物理化学特征的氨基酸替换在生物学上仍可能具有功能,但在序列恢复率指标中受到惩罚。当前最先进的序列设计模型(ProteinMPNN)实现了 52%⁴ 的序列恢复率。我们注意到 ProteinMPNN 除了编码器外还有解码器,而我们的模型只有一个编码器。它们的解码器由教师强制训练,解码顺序随机确定。他们的方法自然比我们的仅编码器模型更具表现力,在该模型中,所有氨基酸身份都是联合预测的,因为自回归解码器可以同时调节结构和(当前解码的)序列信息。
我们还研究了“局部氨基酸环境”的不同定义对模型性能的影响。我们的初始模型遵循 ProteinMPNN 的 K=48 模型(每个残基节点与其最近的 48 个邻居正好有 48 条边)。我们推断,这可能会导致两个空间上接近的残基共享许多共同的邻居,并可能导致过度平滑。由于远离配体的氨基酸不太可能影响靠近配体的氨基酸概率,这也可能使模型更加“以配体为中心”。为了解决这个问题,我们进行了一次扫描,改变 K=32、16、8,并观察到损失和准确度没有显着变化(图 4-5)。我们注意到,随着模型的训练,评估损失增加,而准确度保持相对稳定。某些氨基酸组的化学可互换性可以解释这些趋势。当正确预测氨基酸身份时,模型可能会同时以高 softmax 概率预测化学相似的氨基酸,这会导致更高的交叉熵损失。我们还注意到,我们选择不在每个图卷积层之间使用激活函数,因为根据经验,包括一个激活函数会增加评估损失并降低评估准确性。
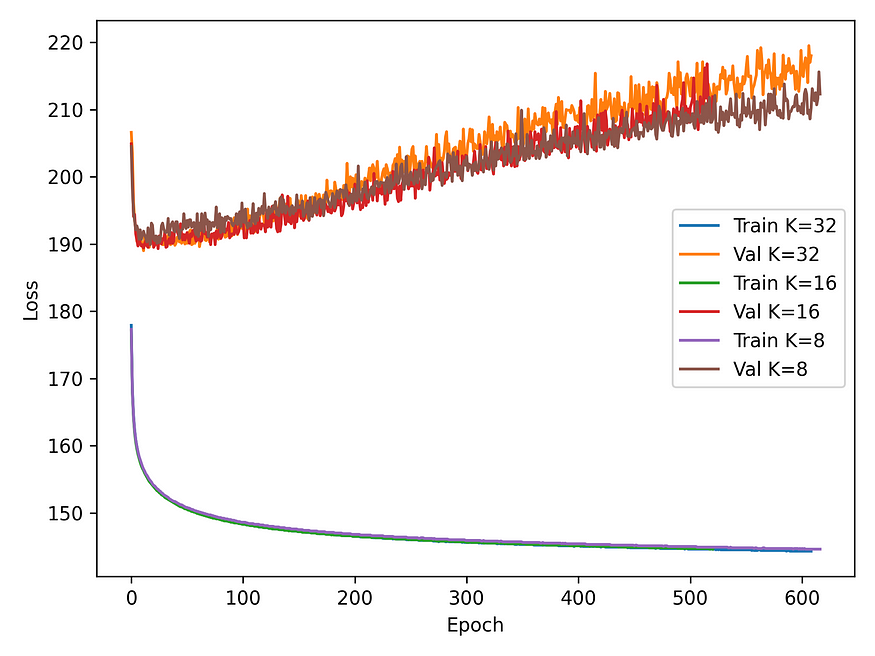
图 3:不同 K 设置下的训练和验证损失曲线(K 个最近邻居与查询/中心节点形成一条边)
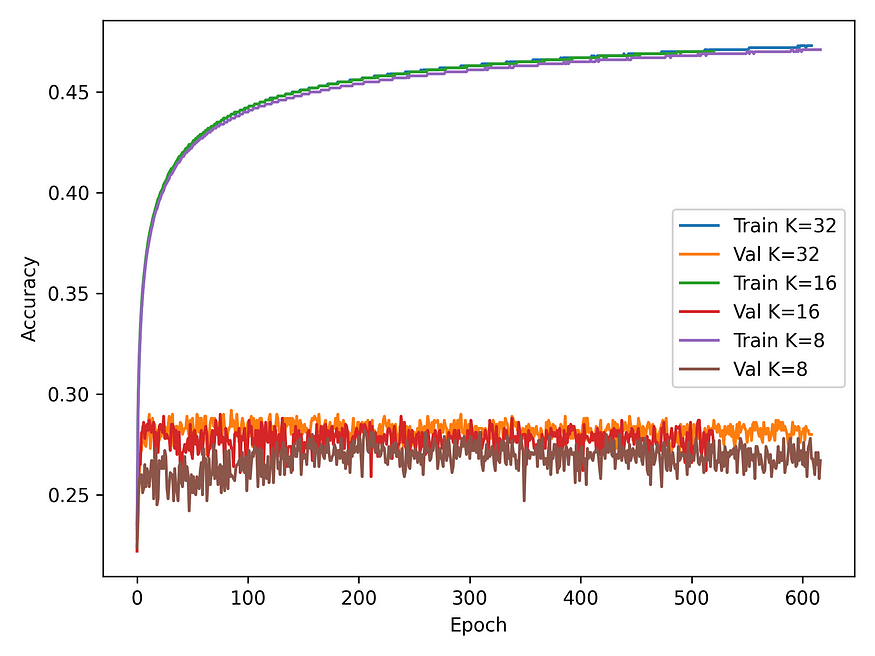
图 4:不同 K 设置(形成边的最近邻)的训练和验证准确度曲线
我们决定在训练期间不使用 QM 特征,因为在推理期间它会限制模型的使用情况——用户需要为每个配体计算 QM 特征,这在计算上非常昂贵。我们推断,对于在许多蛋白质结构中发现的常见配体,缺乏 QM 数据不会显著降低性能。将元素类型编码为节点特征可能足以让模型隐式学习决定蛋白质配体结合口袋中氨基酸环境的化学性质。QM 数据对于预测结合更多奇异配体(例如新型药物)的蛋白质序列最有用,因为这些特征可能允许模型明确学习丰富的化学信息。输入 PDB 中的配体应该是质子化的,因为该模型是用化学有效的质子化分子训练的,但该模型也可以处理非质子化(non-protonated)的配体。
讨论
一个潜在的改进领域可以来自添加更深的解码器模块并对其进行类似于 ProteinMPNN 的训练——如果我们关于解码器重要性的假设成立,我们预计验证准确率会有所提高。更广泛的超参数扫描,例如改变噪声水平(在 ProteinMPNN 中,他们观察到特定噪声水平比其他噪声水平更理想),可能会有所帮助。虽然 MD 模拟旨在用作数据增强,但初步实验并未显示使用完整 MD 数据集与仅使用初始 PDB 结构中的第一帧相比具有显着优势。这一发现的一个潜在原因是 MD 轨迹中的某些帧对应于低概率或高能量状态,导致模型学习可能不切实际的生物物理相互作用。为了更好地利用 MD 数据,我们可以使用均方根误差截止值对帧进行聚类,然后仅在属于人口稠密的簇的 MD 帧上训练模型。这将确保只学习生化上可能的构象,同时仍允许一定的多样性。最后,虽然配体结合蛋白设计的适用性很广,但该模型解决的是一项非常特殊的任务,其中结合物与配体靶标的结构已经确定。为了实现完全的通用性和设计表达性,我们希望找到一种方法,能够同时确定设计蛋白质的序列,以及配体相对于蛋白质结构的位置和构象(conformation)。
GitHub 仓库
https://github.com/tianyu-lu/heterognn
PyG 代码片段
我们利用 GENConv 包装器自动检测输入和输出形状的功能。边属性会自动投影到与节点特征相同的维度中。
class HeteroGNN(torch.nn.Module):
""" Heterogeneous Graph Neural Network for protein design given ligand atomic context"""
def __init__(self, hidden_channels, out_channels, num_layers):
super().__init__() self.convs = torch.nn.ModuleList()for _ in range(num_layers):
conv = HeteroConv({('residue', 'near', 'residue'): GENConv((-1, -1), hidden_channels, edge_dim=5*5*16, expansion=4, aggr='add'),
('residue', 'near', 'atom'): GENConv((-1, -1), hidden_channels, edge_dim=5*16, expansion=4, aggr='add'),
('atom', 'near', 'residue'): GENConv((-1, -1), hidden_channels, edge_dim=5*16, expansion=4, aggr='add'),
('atom', 'near', 'atom'): GENConv((-1, -1), hidden_channels, edge_dim=16, expansion=4, aggr='add'),
}, aggr='sum') self.convs.append(conv) self.bn = nn.BatchNorm1d(hidden_channels) self.lin = Linear(hidden_channels, out_channels)def forward(self, data):
for conv in self.convs: x_dict = conv(data.x_dict, data.edge_index_dict, data.edge_attr_dict)x_dict = {key: self.bn(x) for key, x in x_dict.items()}
return self.lin(x_dict['residue'])
Colab 笔记本演示
https://colab.research.google.com/drive/1x_-gh5zWBCluOha4Z-WrfdzF6EFOt2IB?usp=sharing
作者
Braxton Bell & Tianyu Lu
参考
1) Krishna, R., Wang, J., Ahern, W., Sturmfels, P., Venkatesh, P., Kalvet, I., Lee, G.R., Morey-Burrows, F.S., … & Baker. D. Generalized Biomolecular Modeling and Design with RoseTTAFold All-Atom. bioRxiv (2023).
2) Siebenmorgen, T., Menezes, F., Benassou, S., Merdivan, E., Kesselheim, S., Piraud, M., … & Popowicz, G. M. MISATO-Machine learning dataset of protein-ligand complexes for structure-based drug discovery. bioRxiv (2023)
3) Wang R., Fang, X., Lu, Y., Yang, C. Y., Wang. S. The PDBbind Database: Methodologies and Updates. J. Med. Chem. 48, 4111–4119 (2005).
4) Dauparas, J., Anishchenko, I., Bennett, N., Bai, H., Ragotte, R. J., Milles, L. F., … & Baker, D. Robust deep learning–based protein sequence design using ProteinMPNN. Science. 378, 49–56 (2022).
5) Li, G., Xiong, C., Thabet, A., Ghanem, B. DeeperGCN: All You Need to Train Deeper GCNs. arXiv. (2020).
6) Hu, Weihua, et al. “Strategies for pre-training graph neural networks.” arXiv preprint arXiv:1905.12265 (2019).



